
Abstract
Genome-wide association studies (GWAS) have identified numerous loci associated with Parkinson’s disease. The specific genes and variants that drive the associations within the vast majority of these loci are unknown. We aimed to perform a comprehensive analysis of selected genes to determine the potential role of rare and common genetic variants within these loci. We fully sequenced 32 genes from 25 loci previously associated with Parkinson’s disease in 2,657 patients and 3,647 controls from three cohorts. Capture was done using molecular inversion probes targeting the exons, exon-intron boundaries and untranslated regions (UTRs) of the genes of interest, followed by sequencing. Quality control was performed to include only high-quality variants. We examined the role of rare variants (minor allele frequency < 0.01) using optimized sequence Kernel association tests (SKAT-O). The association of common variants was estimated using regression models adjusted for age, sex and ethnicity as required in each cohort, followed by a meta-analysis. After Bonferroni correction, we identified a burden of rare variants in SYT11, FGF20 and GCH1 associated with Parkinson’s disease. Nominal associations were identified in 21 additional genes. Previous reports suggested that the SYT11 GWAS association is driven by variants in the nearby GBA gene. However, the association of SYT11 was mainly driven by a rare 3’ UTR variant (rs945006601) and was independent of GBA variants (p=5.23E-05 after exclusion of all GBA variant carriers). The association of FGF20 was driven by a rare 5’ UTR variant (rs1034608171) located in the promoter region. The previously reported association of GCH1 with Parkinson’s Disease is driven by rare nonsynonymous variants, some of which are known to cause dopamine-responsive dystonia. We also identified two LRRK2 variants, p.Arg793Met and p.Gln1353Lys, in ten and eight controls, respectively, but not in patients. We identified common variants associated with Parkinson’s disease in MAPT, TMEM175, BST1, SNCA and GPNMB which are all in strong linkage disequilibrium (LD) with known GWAS hits in their respective loci. A common coding PM20D1 variant, p.Ile149Val, was nominally associated with reduced risk of Parkinson’s disease (OR 0.73, 95% CI 0.60-0.89, p=1.161E-03). This variant is not in LD with the top GWAS hits within this locus and may represent a novel association. These results further demonstrate the importance of fine mapping of GWAS loci, and suggest that SYT11, FGF20, and potentially PM20D1, BST1 and GPNMB should be considered for future studies as possible Parkinson’s disease-related genes.
Introduction
Genome-wide association studies (GWASs) are an important tool for identifying genetic associations with complex disorders such as Parkinson’s Disease (Nalls etal., 2014; Chang et al., 2017; Nalls et al., 2019). GWASs examine the association of multiple common variants across the genome with specific traits. Typically, each associated locus includes multiple genes, and very often GWAS alone cannot identify the specific gene or variant within each locus that drives the association. Furthermore, GWASs generally overlook rare variants, while accumulation of rare variants on a specific common allele may drive some of these associations. Even if not located on a specific common allele, rare variants in GWAS genes may contribute to disease. For example, rare variants in GWAS loci genes including SNCA, GBA, LRRK2, VPS13C and GCH1 may cause Mendelian parkinsonism or strongly increase the risk of Parkinson’s disease. (Polymeropoulos et al., 1997; Sidransky et al., 2009; Ran et al., 2016; Emelyanov et al., 2018; Amaral et al., 2019; Paisan-Ruiz etal., 2004; Khan etal., 2005; Lesage et al., 2016; Jansen et al., 2017; Darvish et al., 2018; Schormair et al., 2018; Rudakou et al., 2020; Mencacci et al., 2014; Guella et al., 2015; Lewthwaite et al., 2015; Xu et al., 2017; Yoshino et al., 2018; Rudakou et al., 2019).
Recently, a large-scale GWAS has identified 78 loci associated with risk of Parkinson’s disease (Nalls et al., 2019), yet only for a few of them we have identified the specific genes and variants that drive the association. In some of these loci, there are multiple hits, suggesting that more than one variant, or perhaps more than one gene within the locus, can be associated with Parkinson’s disease. Fine-mapping of GWAS loci may help identifying these genes and variants, as was done for SNCA (Soldner et al., 2016; Krohn et al., 2019b), VPS13C (Lesage et al., 2016; Jansen et al., 2017; Darvish et al., 2018; Schormair et al., 2018; Rudakou et al., 2020) and TMEM175 (Krohn et al., 2020). In the SNCA region, fine mapping has identified independent 5’ and 3’ variants that drive the associations with different synucleinopathies (Krohn et al., 2019b). Similarly, a potentially protective haplotype in VPS13C (Rudakou et al., 2020) and common nonsynonymous variants in TMEM175 have been associated with Parkinson’s Disease through fine mapping, and in the case of TMEM175 also by functional studies (Jinn et al., 2019; Krohn et al., 2020).
In the current study, we fully sequenced 32 genes from 25 different Parkinson’s disease GWAS loci in a total of 2,657 cases and 3,647 controls. We examined the association of common and rare variants with Parkinson’s Disease, including the role of rare bi-allelic variants.
Materials and Methods
Study Population
The study population included, consecutively recruited, unrelated patients with Parkinson’s Disease (n=2,657) and controls (n=3,647) from three cohorts, collected at McGill University (Quebec, Canada and Montpellier, France), Columbia University (New York, NY, USA), and the Sheba Medical Center (Israel). The McGill cohort was recruited in Quebec, Canada (in part with the assistance of the Quebec Parkinson’s Network (Gan-Or etal., 2020)) and in Montpellier, France. It includes 1,026 Parkinson’s Disease patients (mean age 61.5±11.3, 62.6% male) and 2,588 controls (mean age 54.5±14.2, 47.4% male), all unrelated of French-Canadian/French ancestry. The Columbia cohort includes 1,026 Parkinson’s Disease patients (mean age 59.4±11.7, 64.2% male) and 525 controls (mean age 64.2±10.1, 37.1% male). This cohort has been previously described in detail, and is mainly composed of participants with European descent, but 21.2% of Parkinson’s Disease patients and 18.7% controls are of Ashkenazi Jewish (AJ) origin (Alcalay et al., 2016). The Sheba cohort included 605 Parkinson’s Disease patients (mean age 60.7±11.9, 62.3% male) and 534 controls (mean age 34.0±7.0, 55.8% male), all of full AJ origin; previously described in more details (Ruskey et al., 2019). Movement disorder specialists diagnosed Parkinson’s Disease with UK brain bank criteria (Hughes et al., 1992) or the MDS clinical diagnostic criteria (Postuma et al., 2015).
As detailed in the Statistical Analyses subsection, due to the differences in age and sex, statistical analyses were adjusted and included age and sex as co-variates. To account for ethnical heterogeneity of the Columbia cohort, an ethnicity covariate was also introduced for the analysis of this cohort (GWAS data was not available, therefore the reported ethnicity was used and not principal components). All three cohorts were sequenced in the same lab (McGill University), using the same protocol.
Standard Protocol Approvals, Registrations, and Patient Consents
The institutional review boards approved the study protocols and informed consent was obtained from all individual participants before entering the study.
Target genes
A recent Parkinson’s Disease GWAS identified 78 loci, however the current study was designed and performed before its publication and the target genes were taken from earlier literature (Nalls et al., 2014; Chang et al., 2017). Our target genes included: ACMSD, BST1, CCDC62, DDRGK1, FGF20, GAK, GCH1, GPNMB, HIP1R, INPP5F, ITGA8, LAMP3, LRRK2, MAPT, MCCC1, PM20D1, RAB25, RAB29, RIT2, SCARB2, SETD1A, SIPA1L2, SLC41A1, SNCA, SREBF1, STK39, STX1B, SYT11, TMEM163, TMEM175, USP25 and VPS13C. These 32 genes included three known Parkinson’s disease related genes (LRRK2, GCH1, SNCA) found in GWAS loci in order to perform full sequencing assessment. The other 29 genes were selected for sequencing based on assessment that included effects of quantitative trait loci, brain expression, potential association with known Parkinson’s disease-causing genes and involvement in pathways potentially involved in Parkinson’s disease, such the autophagy-lysosomal pathway, mitochondria quality control and endolysosomal recycling.
Sequencing
The coding regions, exon-intron boundaries and the 5’ and 3’ untranslated regions (UTRs) of the genes listed above were targeted using molecular inversion probes (MIPs), as previously described (O’Roak et al., 2012). All MIPs used to sequence the genes of the study are detailed in Supplementary Table 1. Targeted DNA capture and amplification was done as previously described (Ross et al., 2016), and the full protocol is available upon request. The library was sequenced using Illumina HiSeq 2500/4000 platform at the McGill University and Genome Quebec Innovation Centre. Sanger sequencing was performed for rare variants with significant association to Parkinson’s disease. Primers and conditions are available upon request.
Data processing and quality control
Reads were mapped to the human reference genome (hg19) with Burrows-Wheeler Aligner (Li and Durbin, 2009). Post-alignment quality control and variant calling was performed using Genome Analysis Toolkit (GATK, v3.8) (McKenna et al., 2010). Variant annotation was done with ANNOVAR (Wang et al., 2010). Data on the frequency of each variant in different populations were extracted from the public database Genome Aggregation Database (GnomAD) (Lek et al., 2016).
The quality control pipeline that was used in this study is available on GitHub (https://github.com/gan-orlab/MIPVar.git). All calls with less than 25% of the reads containing the observed allele have been removed. The remaining quality control (QC) was performed using GATK and PLINK software v1.9 (Purcell et al., 2007). Genotyping quality (GQ) < 30 was used as a threshold for variant exclusion. Different depths of coverage thresholds were used for common and rare variants. Common variants threshold for inclusion was ≥ 15x, while rare variants (minor allele frequency [MAF] < 0.01) thresholds were more stringent and repeated twice, at ≥ 30x and ≥ 50x. Variants with genotype rate lower than 90% were excluded. Variants that deviated from Hardy-Weinberg equilibrium set at p < 0.001 were filtered out. Threshold for missingness difference between cases and controls was set at p = 0.05 and the filtration script adjusted it with Bonferroni correction. Samples with average genotyping rate of less than 90% were excluded.
Statistical analysis
The burden of rare variants in each of the target genes was tested using the optimized sequence Kernel association test (SKAT-O, R package) (Lee et al., 2012). In addition, we examined the burden of specific variant subgroups including: i) Funct.: Potentially functional included all nonsynonymous mutations, stop gain/loss, frameshift mutations, splicing variants located within two base pairs of exon-intron junctions, and variants that were flagged by ENCODE (explained below). ii) NS: nonsynonymous variants. iii) LOF: loss-of-function variants included stop gain/loss, frameshift, and splicing variants located within two base pairs of exon-intron junctions. iv) CADD: variants with the Combined Annotation Dependent Depletion (CADD) score ≥ 12.37; this threshold indicates the top 2% variants predicted to be the most damaging in the genome (Amendola et al., 2015). v) ENCODE: variants that are predicted to have regulatory activity, such as promoter, enhancer, activator and repressor, based on the chromatin profiling studies (Ernst et al., 2011). To increase power, rare variants were assessed in the three merged cohorts (since rare variants are less affected by ethnicity). Before merging, allele frequencies were compared between the control groups and between the patient groups. We have also compared allele frequencies between young controls and elderly controls specifically. Since there were no significant differences when comparing the control groups and the patient groups, we could merge them for the analysis. Analysis of the individual cohorts was also performed post-hoc. Similarly to exome-wide burden analyses of rare variants (Kenna et al., 2016), Bonferroni correction for statistical significance was performed based on the number of genes that were tested, and the thresholds are p < 1.85E-03 for genes with variant coverage of ≥ 50x and p < 1.56E-03 at ≥ 30x. Since the different categories and coverages are not independent, we have also applied false discovery rate (FDR) correction to account for all analyses in all categories combined. To determine if an identified association was mainly driven by a single variant, we performed a ‘leave-one-out’ analysis, in which we systematically excluded a single variant each time for a certain gene and examine whether excluding this variant affected the observed results. Variants that their exclusion led to non-significant or to a highly reduced statistical significance were considered as main drivers of the initially observed association.
The association between common variants and Parkinson’s Disease was examined by logistic regression models using PLINK v1.9, with the status (patient or control) as a dependent variable, age and sex as covariates in all cohorts. Ethnicity as an additional covariate in the Columbia University cohort due to the ethnical heterogeneity. Meta-analysis of our three cohorts was conducted using METAL (R package) (Willer et al., 2010) with a fixed effect model. Cochran’s Q-test was applied to test for residual heterogeneity. LD of common variants and their respective GWAS top marker was examined using the LDlink online tool LDmatrix (https://ldlink.nci.nih.gov/?tab=ldmatrix) (Machiela and Chanock, 2015). Effects of common and rare variants on expression levels (eQTL) or splicing (sQTL) was evaluated using GTEx Portal online tool (https://www.gtexportal.org/home/).
Data availability
Anonymized data is available upon request by any qualified investigator.
Results
Coverage and variants identified
The average coverage for all 32 genes included in the current analysis was 642X, with a range of 86-1,064 (median 688) across all genes. An average of 97% of the targeted regions was covered at ≥ 15X, 96% at ≥ 30X, and 92% at ≥ 50X. Supplementary Table 2 details the average coverage and the percent coverage for each gene. At coverage of ≥ 30X, we found 3,327 rare variants and at > 50X, 1,713 rare were identified (Supplementary Tables 3 and 4). A total of 311 common variants were identified across all genes in all three cohorts (Supplementary Table 5) with a coverage of ≥15X.
Rare variants in SYT11, FGF20 and GCH1 are associated with Parkinson’s disease
Table 1 details the SKAT-O results of the analysis in the three combined cohorts, and Supplementary Table 6 details the results per cohort. After Bonferroni correction, SYT11, FGF20 and GCH1 were associated with Parkinson’s Disease. Nominal associations were identified in 21 genes (Table 1, Supplementary Table 6). When considering all variants with MAF < 0.01 in SYT11, there was a strong association with Parkinson’s Disease (p = 5.3E-06, Table 1, FDR-corrected p=0.0017, Supplementary Table 7), mainly driven by a SYT11 3’ UTR variant (rs945006601) with MAF of 0.004 in Parkinson’s Disease patients and 0.00015 in controls (OR = 27.0, 95%CI 3.6-202.6, p = 0.0013). This association was identified at a coverage of ≥ 50X, which provides high certainty that rare variants are being called correctly. We further sequenced this variant using Sanger sequencing, and all carriers have been confirmed, as well as randomly selected non-carriers. At 30X, the association was weaker (p=0.02, Table 1), possibly due to inclusion of other variants with coverage of between 30-50X that diluted the association. It was previously reported that variants in the SYT11 locus are in LD with known GBA variants such as p.Glu326Lys and p.Asn370Ser, and that the association seen with SYT11 is driven by the GBA Variants (Blauwendraat et al., 2018). However, the rs945006601 SYT11 variant that drives the association in the current study is not in LD with these variants, but with the wild-type GBA allele. To further test if the identified association in SYT11 is driven by other GBA variants, we removed carriers of all GBA variants that were ever reported to be potentially pathogenic, and the association of SYT11 with PD remained statistically significant (p=5.23E-05).
When analyzing FGF20, a burden of all rare variants was demonstrated (p=0.0002, Table 1, FDR-corrected p=0.026), mainly driven by a rare 5’ UTR variant, rs1034608171, which is located in the promoter region of the gene, with MAF of 0.002 in Parkinson’s Disease patients and 0.00015 in controls (OR = 13.4, 95% CI 1.7-106.2, p = 0.014). This association was discovered at a coverage of ≥ 30X, but at ≥ 50X it was not identified since the average coverage of this variant was below 50X. However, the variant was confirmed using Sanger sequencing.
The association of GCH1 with Parkinson’s Disease is driven by rare nonsynonymous variants (Supplementary Tables 3 and 4), some of which are known to cause dopamine responsive dystonia as we have previously reported (Rudakou et al., 2019). We repeated the analyses above focusing only on early onset PD patients with age at onset <50 years (n=517). We did not identify any associations at coverages of ≥ 30X or ≥ 50X (Supplementary Tables 8 and 9, respectively), yet this is an underpowered analysis, and studies in larger cohorts of early onset PD are necessary.
After removing the LRRK2 p.Gly2019Ser variant from the analysis (a total of 179 carriers, 10 carriers form the McGill cohort, 82 from the Columbia cohort and 87 in Sheba cohort), there was no statistically significant burden of other rare LRRK2 variants in our cohorts. However, we did identify two variants, p.Arg793Met (rs35173587) and p.Gln1353Lys (rs200526782), in ten and eight controls, respectively, but not in Parkinson’s Disease. Neither of these two variants had a statistically significant association after Bonferroni correction.
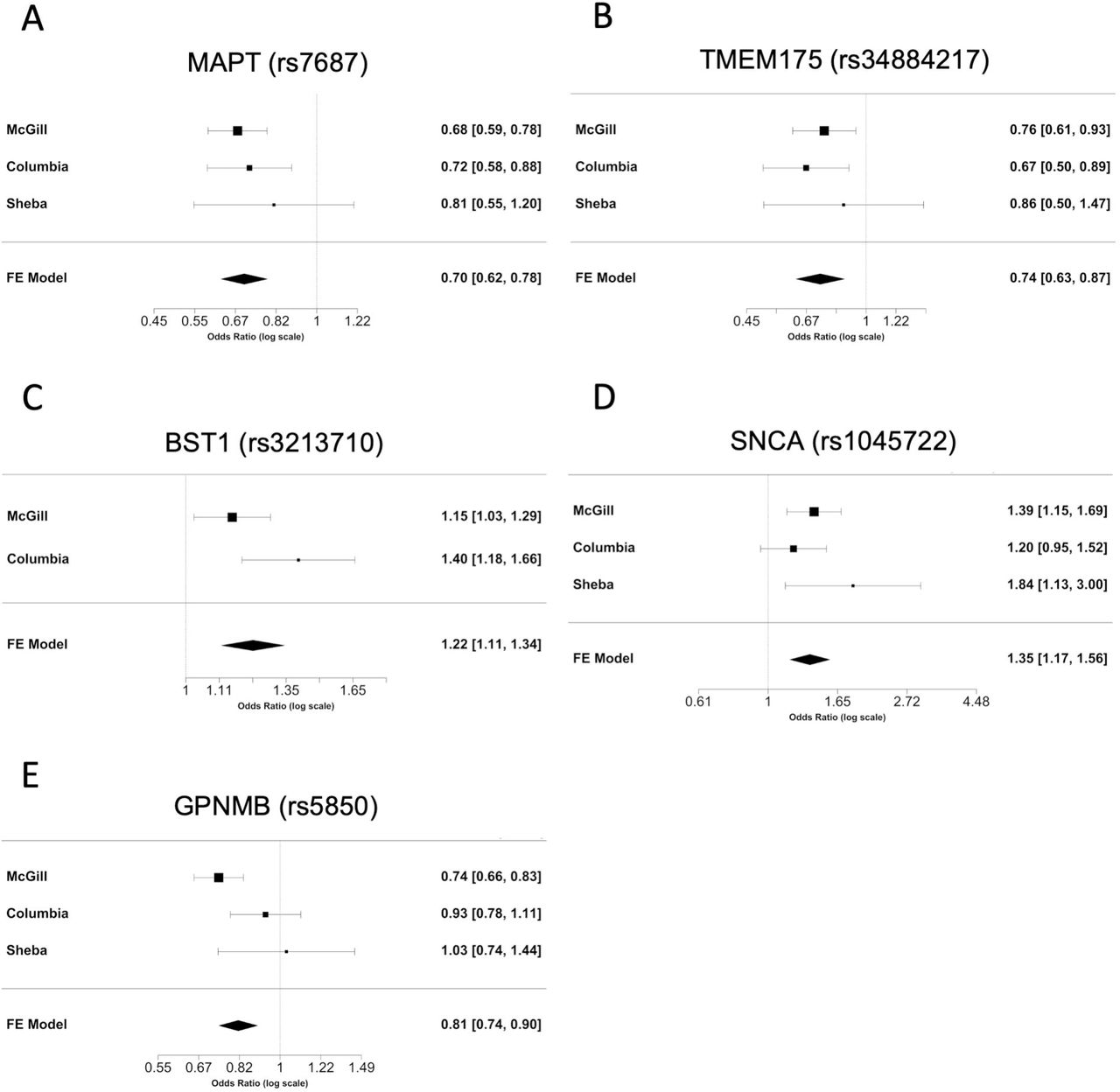
Association of common variants with potential functional effects in known Parkinson’s disease loci
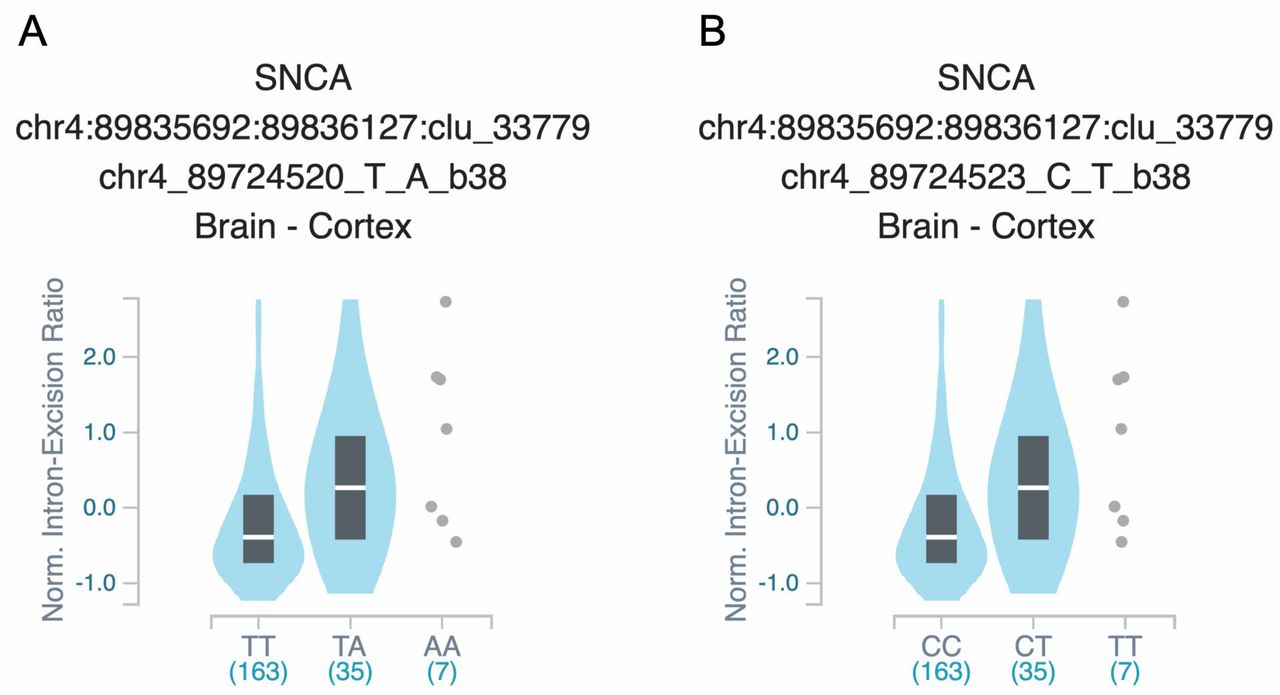
To examine the association of common variants in the 32 genes, we performed logistic regression adjusted for age and sex in all three cohorts, and also adjusted for ethnicity in the Columbia University cohort. Additionally, our three cohorts were meta-analyzed. After Bonferroni correction we found 24 variants from five genes associated with risk of Parkinson’s Disease (Table 2). Forest plots in Fig. 1 show the meta-analysis results for the tagging variants of these five genes. We further examined the LD of these variants with the known Parkinson’s Disease-associated variants from the most recent Parkinson’s Disease GWAS, and their potential functional effects (detailed in Methods) in ENCODE, GTEx and UCSC genome browser. In the MAPT locus, 17 common variants at full LD, all represent the MAPT H2 haplotype, were associated with reduced risk of Parkinson’s Disease (Table 2). The variants identified in TMEM175, BST1, SNCA and GPNMB are all in strong LD with known GWAS hits in their respective loci (Table 2). Two SNCA 3’ UTR variants were in full LD between themselves, and in strong LD with rs356219, a known 3’ GWAS hit in the SNCA locus. These two variants are located within a strong enhancer region and are associated with altered splicing of SNCA in the

Each panel presents the individual odds ratio and 95% confidence interval for each cohort, and the results of the meta-analysis for the tagging variant of six loci identified in our study. Meta-analysis p-values: A. MAPT (p=8.28E-10); B. TMEM175 (p=2.11E-04); C. BST1 (p=3.14E-05); D. SNCA (p=4.08E-05); E. GPNMB (p=2.19E-05)
Cortex (Fig. 2). Three GPNMB variants were identified, one of which (rs5850) is located at the 3’ UTR of GPNMB and associated with reduced expression of GPNMB in numerous brain regions including the substantia nigra, putamen and cortex (Supplementary Fig. 1). A coding variant in PM20D1, p.Ile149Val (rs1891460), was nominally associated with reduced risk of Parkinson’s Disease (OR = 0.73, 95% CI 0.60-0.89, p = 1.161E-03, specific frequencies in each cohort are in Supplementary Table 5). This variant is located within the PARK16 region which also includes RAB29 (previously named RAB7L1), SLC41A1 and other genes. However, this variant is not in LD with the top GWAS hits within this locus (r2 = 0.04, D’ = 0.46).

{kind=link}
{kind=link}
Figure is processed from GTeX (https://www.gtexportal.org), and represent the splicing quantitative trait locus (sQTL) effects of: A. rs1045722 in the 3’ UTR of SNCA and B. rs3857053 in the 3’ UTR of SNCA
Discussion
In the current study, we examined the role of common and rare variants in 32 genes located within known Parkinson’s Disease risk loci, by performing targeted next generation sequencing of the coding and 5’ and 3’ untranslated regions, followed by association tests. We found a statistically significant burden, after Bonferroni correction, of rare variants in SYT11, FGF20 and GCH1, and nominal associations (p<0.05) with other genes and variants (Table 1, Supplementary Tables 3,4,6). In addition, we have identified common coding and putative regulatory variants associated with Parkinson’s Disease in MAPT, TMEM175, BST1, SNCA and GPNMB, including a potential novel association with a nonsynonymous variant in PM20D1, p.Ile149Val, which is nominally significant and requires further confirmation.
Three genes, SYT11, FGF20 and GCH1 were identified with a statistically significant burden of rare variants comparing Parkinson’s Disease to controls. SYT11 is located close to the GBA locus, and it was reported that the GWAS signal from SYT11-GBA locus in Parkinson’s Disease can be attributed to coding GBA variants (Blauwendraat et al., 2018). The SYT11 variant mainly responsible for the association in our data is in weak LD with two coding GBA variants, p.Glu326Lys (p.E326K) and p.Asn370Ser (p.N370S). After exclusion of carriers of these variants, and further exclusion of all other GBA variants, the association of SYT11 remained strong. These results suggest that SYT11 may independently be associated with risk of PD. The Protein encoded by SYT11, Synaptotagmin 11, belongs to the synaptotagmin family, known calcium sensors mediating calcium-dependent regulation of membrane trafficking in synaptic transmission (Xie et al., 2017). Synaptotagmin 11 is involved in regulation of endocytosis and vesicle recycling process, which is essential for neurotransmission (Wang et al., 2016; Xie et al., 2017). Overexpression of Synaptotagmin 11 in mice led to impairment of DA transmission (Wang et al., 2018). Synaptotagmin 11 was also reported to be a physiological substrate for E3 ubiquitin ligase – parkin, playing a critical role in parkin-related neurotoxicity (Wang et al., 2018). Involvement of Synaptotagmin 11 in other mechanisms potentially related to Parkinson’s Disease such as autophagy-lysosomal pathway (Bento et al., 2016), phagocytosis of α-synuclein fibrils (Du et al., 2017), and repair of injured astrocytes via lysosome exocytosis regulation (Sreetama et al., 2016), suggests that SYT11 needs to be considered as a target for further research in PD. Fibroblast growth factor 20 (FGF20) encodes a neurotrophic factor preferentially expressed in the substantia nigra pars compacta. It regulates central nervous system development and function (International Parkinson Disease Genomics Consortium, 2011) and plays a role in dopaminergic neurons differentiation and survival (Itoh and Ohta, 2013). There is a number of conflicting reports about FGF20 overexpression increasing α-synuclein levels in dopaminergic neurons (Wang et al., 2008; Wider et al., 2009; Sekiyama et al., 2014; Tarazi et al., 2014). The rare variant (rs1034608171) that drives the association in our data is located in a regulatory region and its potential functional effects would require additional studies. GCH1 encodes GTP-cyclohydrolase 1, an essential enzyme for the dopamine synthesis pathway in the nigrostriatal cells (Kurian et al., 2011). Rare GCH1 variants may cause DOPA-responsive dystonia, and the same variants have also been associated with Parkinson’s Disease (Mencacci et al., 2014; Rudakou et al., 2019). Since these mutations are rare, they only reached statistical significance in the McGill cohort, as we have previously described (Rudakou et al., 2019). The two LRRK2 variants, p.Arg793Met and p.Gln1353Lys, that were found only in controls require additional studies to examine their potential protective roles in Parkinson’s Disease. LRRK2 is a well-validated Parkinson’s Disease related gene (Bandres-Ciga et al., 2020).
All 17 MAPT variants that were found to be significantly associated with Parkinson’s Disease are in perfect LD with each other, and represent the H2 haplotype, known to be associated with reduced risk of Parkinson’s Disease (Li et al., 2018). The identified coding variants in MAPT are located within exons that are likely not expressed in the CNS (Caillet-Boudin et al., 2015), suggesting that the association in this locus may be due to regulatory variants. TMEM175 is located in the fourth most significant GWAS locus in Parkinson’s Disease (Nalls et al., 2019), and encodes a transmembrane potassium channel responsible for K+ conductance in endosomes and lysosomes (Cang et al., 2015). TMEM175 regulates lysosomal function through maintenance of lysosomal membrane potential and luminal pH stability, as well as its role in autophagosome-lysosome fusion (Lee et al., 2017). In a recent genetic and functional study, two coding TMEM175 variants, including the variant identified here, were shown to be associated with risk of Parkinson’s Disease, risk of REM-sleep behavior disorder, and glucocerebrosidase activity (Krohn et al., 2019a).
The BST1 and SNCA variants associated with risk of Parkinson’s Disease in the current study are in strong LD with the known GWAS markers in their respective loci. The BST1 variant is intronic without known function, and the SNCA 3 ’UTR variants are located within strong SNCA enhancers. Whether these SNCA variants drive the association with Parkinson’s Disease and their potential functional effects require additional studies. GPNMB encodes the glycoprotein nonmetastatic melanoma protein B a type 1, which is a transmembrane glycoprotein shown to have a role in melanoma (Taya and Hammes, 2018). Patients with melanoma have an increased risk of Parkinson’s Disease (Liu et al., 2011), and Parkinson’s Disease patients have an increased risk for melanoma (Bertoni et al., 2010). GPNMB may have a role in the lysosome (Tomihari et al., 2009) and in regulation of microglial inflammation (Dzamko et al., 2015; Herrero et al., 2015). While the expression of GPNMB in brain is overall low, the two variants identified in the current study are associated with reduced GPNMB expression in various brain tissues (Supplementary Fig. 1). One of the variants, rs5850, is located within the 3’ UTR of GPNMB, but there is no clear regulatory function for this variant.
Peptidase M20 Domain Containing 1 (PM20D1) encodes a protein that regulates the production of N-fatty-acyl amino acids which have a function of chemical uncouplers of mitochondrial respiration (Long et al., 2016). Mitochondrial quality control is likely important in Parkinson’s Disease, and genes involved in mitophagy are involved in familial forms of Parkinson’s Disease (Park et al., 2018). PM20D1 is located within the PARK16 locus which also contains SLC45A3, NUCKS1, RAB29 (previously named RAB7L1) and SLC41A1 (Satake et al., 2009; Simon-Sanchez et al., 2009; Nalls et al., 2019).Several studies have highlighted RAB29 as the potential gene associated with Parkinson’s Disease in this locus (Gan-Or et al., 2012),as it was shown to interact with LRRK2 (MacLeod et al., 2013) and affect lysosomal function (Eguchi et al., 2018). In the current study we identified a common nonsynonymous variant in PM20D1, p.Ile149Val (rs1891460), which is not in LD with the Parkinson’s Disease GWAS marker in this locus. Since this variant is not reported in ClinVar, has a low CADD score (1.01), and the statistical association is of borderline significance when correcting by the number of genes, it is possible that this association was found by chance. Replications in other cohorts will be required to determine whether this variant is associated with Parkinson’s Disease.
The associations described above were detected using different coverage thresholds for the detection of common and rare variants. For common variants, we used a threshold of coverage of ≥15X, which based on our previous studies (Rudakou et al., 2019; Krohn et al., 2019b; Rudakou et al., 2020; Krohn et al., 2020) is highly reliable for detection of known common variants. Here too, when comparing to available data from GWAS genotyping, we were able to confirm with 100% accuracy the presence of common variants in MAPT, TMEM175 and GPNMB in samples with available data. The different thresholds of ≥ 30X and ≥ 50X are being used to increase our confidence that the variants and the results are real. The ≥ 50X threshold is highly reliable, yet also at ≥ 30X most detected variants are real. In the current study, the variants driving the associations in FGF20 and GCH1 has been fully confirmed with Sanger sequencing, further validating our results.
Our study has several limitations. The Parkinson’s disease patients and controls are not matched by sex and age, hence we adjusted for these variables in the statistical analysis. We found no significant difference between allele frequencies of young (<40 years old) and old controls, which allowed us to use all of the controls in our analyses. In addition, the coverage of some of the genes we analyzed was suboptimal (Supplementary Table 2), and we did not target intronic regions and other potentially regulatory regions outside the 5’ and 3’ UTRs, which may also affect gene function. Whole genome sequencing studies will be required to study these regions and overcome the coverage limitations. Another limitation of our study is the potential genetic heterogeneity, since cohorts that with both European or Ashkenazi Jewish patients and controls were included in the analyses. While we did include ethnicity as a covariate when needed, the Ashkenazi Jewish ancestry was a reported ancestry, not confirmed by GWAS data. Therefore, there could still be some residual effect of ethnicity, and our results should be confirmed in additional populations. Of note, when we removed the Ashkenazi Jewish individuals from the analysis, we yielded similar results, yet less significant due to the reduced power.
Our results, combined with previous studies, highlight the importance of performing fine mapping of GWAS loci to identify the variants and genes within each locus that drive the reported associations. Thus far, the causative genes within most Parkinson’s Disease GWAS loci are unknown, with the exception of SNCA, GBA, LRRK2, and likely VPS13C, MAPT, TMEM175 and GCH1. The current study suggests that SYT11, FGF20, and potentially PM20D1, BST1 and GPNMB should be considered as possible Parkinson’s Disease-related genes and targets for further research.
Data Availability
Anonymized data is available upon request by any qualified investigator.
Funding
This work was financially supported by grants from the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives initiative (HBHL), and Parkinson Canada. The Columbia University cohort is supported by the Parkinson’s Foundation, the National Institutes of Health (K02NS080915, and UL1 TR000040) and the Brookdale Foundation.
Competing interests
Ziv Gan-Or has received consulting fees from Lysosomal Therapeutics Inc., Idorsia, Prevail Therapeutics, Denali, Ono Therapeutics, Deerfield and Inception Sciences (now Ventus). None of these companies were involved in any parts of preparing, drafting and publishing this review. Cheryl H. Waters disclosed the following: Research support (Biogen, Roche, Sanofi); consulting fees (Acorda, Amneal, Impel, Kyowa, Mitsubishi, and Neurocrine); speaker’s honoraria (Acadia, Acorda, Adamas, Amneal, and US WorldMeds)
Supplementary material
Supplementary Table 1: Detailed information on molecular inversion probes (MIPs)
Supplementary Table 2: Coverage statistics
Supplementary Table 3: All identified rare variants with 30x minimum depth of coverage
Supplementary Table 4: All identified rare variants with 50x minimum depth of coverage
Supplementary Table 5: All identified common variants
Supplementary Table 6: SKAT-O results for individual cohorts
Supplementary Table 7: FDR-corrected p values of all SKAT-O results at 30x and 50x minimal depth of coverage
>Supplementary Table 8: Results of SKAT-O analysis of early onset PD patients at coverage >=30X
>Supplementary Table 9: Results of SKAT-O analysis of early onset PD patients at coverage >=50X
>Supplementary Figure 1: The 3’UTR GPNMB variant (rs5850) is associated with reduced expression in numerous brain regions.
Acknowledgements
We thank the participants for contributing to the study. GAR holds a Canada Research Chair in Genetics of the Nervous System and the Wilder Penfield Chair in Neurosciences. EAF is supported by a Foundation Grant from the Canadian Institutes of Health Research (FDN grant – 154301). ZGO is supported by the Fonds de recherche du Québec - Santé (FRQS) Chercheurs-boursiers award, in collaboration with Parkinson Quebec, and by the Young Investigator Award by Parkinson Canada. The access to part of the participants for this research has been made possible thanks to the Quebec Parkinson’s Network (http://rpq-qpn.ca/en/). We thank Daniel Rochefort, Helene Catoire, Sandra B. Laurent, Clotilde Degroot and Vessela Zaharieva for their assistance.
Footnotes
Additional author was included. Added page 11 line 21 - page 12 line 2 to clarify the comparison of young and elderly controls. Added a section about potential genetic heterogeneity due to different ethnical origins to the limitations , page 21 lines 15-22. Added clarifications for the difference in results seen in 30x coverage vs. 50x coverage : page 13 line 18 - page 14 line 3; page 14 lines 11-16. Added a brief discussion about the issue of using different depths of coverage : page 20 line 20 - page 21 line 7. Modified the text to better explain the selection of target genes : page 9 lines 14-20. Added a section to better explain the identification of the main drivers of the associations : age 12 lines 8-12. Added the number of LRRK2 G2019S carriers that were removed for the analysis : page 15 lines 1-3. Added a section about FDR correction In the Methods: page 12 lines 6-8. Added information on the validations of significant results using another genotyping technology: rare SYT11 variants page 13 line 18 - page 14 line 2, rare FGF20 variants page 14 lines 11-16, common variants in page 20 line 20 - page 21 line 3. Corrected the text related to LD of identified SYT11 variant with GBA page 14 lines 3-10. Figure 3 was moved to the supplementary materials. Supplemental files updated
Abbreviations
- AJ
- Ashkenazi Jewish
- CADD
- Combined Annotation Dependent Depletion
- CI
- confidence interval
- CNS
- central nervous system
- DOPA
- dihydroxyphenylalanine
- DP
- depth of coverage
- eQTL
- expression quantitative trait loci
- GATK
- Genome Analysis Toolkit
- GnomAD
- Genome Aggregation Database
- GTEx
- Genotype-tissue expression
- GWAS
- Genome-wide association study
- hg19
- human genome version 19
- LD
- linkage disequilibrium
- LOF
- loss-of-function
- MAF
- minor allele frequency
- MIPs
- molecular inversion probes
- NS, Nonsyn
- non-synonymous
- OR
- odds ratio
- QC
- quality control
- REM
- Rapid Eye Movement
- SKAT-O
- optimized sequence kernel association test
- sQTL
- splicing quantitative trait loci
- UTR
- untranslated region
- yo
- years of age
References



