
ABSTRACT
Background Studies in humans and experimental models highlight a role of interleukin-6 (IL-6) in cardiovascular disease. Indirect evidence suggests that inhibition of IL-6 signaling could lower risk of coronary artery disease. However, whether such an approach would be effective for ischemic stroke and other cardiovascular outcomes remains unknown.
Methods In a genome-wide association study (GWAS) of 204,402 European individuals, we identified genetic proxies for downregulated IL-6 signaling as genetic variants in the IL-6 receptor (IL6R) locus that were associated with lower C-reactive protein (CRP) levels, a downstream effector of IL-6 signaling. We then applied two-sample Mendelian randomization (MR) to explore associations with ischemic stroke and its major subtypes (large artery stroke, cardioembolic stroke, small vessel stroke) in the MEGASTROKE dataset (34,217 cases and 404,630 controls), with coronary artery disease in the CARDIoGRAMplusC4D dataset (60,801 cases and 123,504 control), and with other cardiovascular outcomes in the UK Biobank (up to 321,406 individuals) and in phenotype-specific GWAS datasets. All effect estimates were scaled to the CRP-decreasing effects of tocilizumab, a monoclonal antibody targeting IL-6R.
Results We identified 7 genetic variants as proxies for downregulated IL-6 signaling, which showed effects on upstream regulators (IL-6 and soluble IL-6R levels) and downstream effectors (CRP and fibrinogen levels) of the pathway that were consistent with pharmacological blockade of IL-6R. In MR, proxies for downregulated IL-6 signaling were associated with lower risk of ischemic stroke (Odds Ratio [OR]: 0.89, 95%CI: 0.82-0.97) and coronary artery disease (OR: 0.84, 95%CI: 0.77-0.90). Focusing on ischemic stroke subtypes, we found significant associations with risk of large artery (OR: 0.76, 95%CI: 0.62-0.93) and small vessel stroke (OR: 0.71, 95%CI: 0.59-0.86), but not cardioembolic stroke (OR: 0.95, 95%CI: 0.74-1.22). Proxies for IL-6 signaling inhibition were further associated with a lower risk of myocardial infarction, aortic aneurysm, atrial fibrillation and carotid plaque.
Conclusions We provide evidence for a causal effect of IL-6 signaling on ischemic stroke, particularly large artery and small vessel stroke, and a range of other cardiovascular outcomes. IL-6R blockade might represent a valid therapeutic target for lowering cardiovascular risk and should thus be investigated in clinical trials.
What is new
We identified genetic proxies for downregulated IL-6 signaling that had effects on upstream and downstream regulators of the IL-6 signaling pathway consistent with those of pharmacological IL-6R blockade
Genetically downregulated IL-6 signaling was associated with a lower risk of ischemic stroke, and in particular large artery and small vessel stroke
Similar associations were obtained for a broad range of other cardiovascular outcomes
What are the clinical implications
Inhibition of IL-6 signaling is a promising therapeutic target for lowering risk of stroke and other cardiovascular outcomes and should be further investigated in clinical trials
INTRODUCTION
Stroke is the leading cause of adult disability and the second most common cause of mortality worldwide1, 2 with an increasing burden on global health.3, 4 Inflammation is involved in the pathogenesis of ischemic stroke, as has specifically been demonstrated for large artery atherosclerotic stroke.5, 6 Cytokines regulate inflammatory responses5 and could thus serve as targets for cardiovascular disease prevention.7 In the recent Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS), treatment with an interleukin-1β (IL-1β) antagonist reduced cardiovascular event rates in patients with a history of myocardial infarction.8 However, whether interfering with other cytokines would likewise offer benefit remains largely unknown. Also, there are few data on stroke and other cardiovascular outcomes beyond coronary artery disease.9-11
Interleukin-6 (IL-6), a key regulator of the inflammatory cascade, acts by binding to either its membrane-bound or soluble receptor (IL-6R) and induces proinflammatory downstream effects including increases in the levels of C-reactive protein (CRP).12, 13 IL-6 has been implicated in the pathogenesis of multiple inflammatory diseases and inhibitors of IL-6R are used for the treatment of rheumatoid arthritis,14 inflammatory bowel disease,15 and other autoimmune disorders.16 Downregulation of IL-6 signaling has further been proposed as a potential strategy for lowering cardiovascular risk.11, 13 IL-6 levels have consistently been associated with risk of coronary artery disease in cohort studies.17, 18 Mendelian randomization (MR) studies further showed that a variant in the gene encoding IL-6R with effects resembling pharmacological IL-6R inhibition is associated with a lower risk of coronary artery disease.19, 20 Finally, secondary analyses from CANTOS demonstrated that the magnitude of the therapeutic benefit of IL-1β targeting was associated with the reduction of circulating IL-6 levels11, 21 and that even after IL-1β inhibition, the residual cardiovascular risk was proportional to the post-treatment IL-6 levels.22 These results provide indirect clinical evidence that interfering with IL-6 signaling might lower cardiovascular risk and suggest that an approach directly targeting IL-6 signaling could offer additional benefit for cardiovascular prevention beyond IL-1β inhibition.
The effects of IL-6 signaling on risk of ischemic stroke remain largely unknown. While population-based cohort studies have found that circulating IL-6 levels are associated with a higher risk of ischemic stroke,23, 24 these associations preclude conclusions about causal relationships because of possible confounding and reverse causation bias.25 Also, there are no data on etiological stroke subtypes and other cardiovascular outcomes beyond coronary artery disease. Developing meaningful strategies for the prevention of ischemic stroke and cardiovascular disease in general would require defining these relationships.26
By using genetic variants as proxies for a trait of interest, MR overcomes key limitations of observational studies such as confounding and reverse causation and allows for investigation of causal effects on outcomes.27, 28 MR further allows for prediction of the effects of pharmacological interventions by using variants located close to genes encoding candidate drug targets.29, 30 Hence, MR has become a powerful strategy to prioritize interventions for exploration in clinical trials.28
Here, leveraging data from large genome-wide association studies (GWASs)31-33 and applying MR analyses, we aimed to: (i) identify genetic proxies for downregulated IL-6 signaling on the basis of their effects on CRP levels, a well-established IL-6 signaling downstream effector,13, 20, 34 (ii) validate their utility by comparing the consistency of their effects on upstream regulators and downstream effectors of the IL-6 signaling pathway with the effects of pharmacological IL-6R inhibition, as derived from clinical trials, (iii) explore associations of genetic predisposition to downregulated IL-6 signaling with the risk of ischemic stroke and coronary artery disease, (iv) examine associations with major etiological subtypes of ischemic stroke (large artery, cardioembolic, and small vessel stroke), and (v) examine associations with a broad range of other cardiovascular phenotypes. To derive clinically meaningful effect sizes that would be comparable to those derived from potential future clinical trials, we weighted our instruments based on the CRP-decreasing effects of tocilizumab, a monoclonal antibody targeting IL-6R.
METHODS
Selection of genetic proxies for IL-6 signaling and validation of the instruments
The data sources for this study are described in Table 1. To identify instruments for genetic predisposition to downregulated IL-6 signaling, we selected variants within or near the IL6R gene, which encodes the receptor of IL-6. Specifically, we selected single-nucleotide polymorphisms (SNPs) in the IL6R gene or a region of 300 kB upstream or downstream from the IL6R gene (GRCh37/hg19 coordinates: chr1:154,077,669-154,741,926; Supplementary Figure 1) that were associated with circulating CRP levels. We selected and weighted genetic instruments for genetic predisposition to IL-6 signaling on the basis of their associations with CRP levels, because elevated CRP levels are a well-described downstream effect of IL-6 signaling (Figure 1).13, 20, 34 Genetic association estimates with circulating CRP levels were obtained from a GWAS of 204,402 individuals of European ancestry drawn from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Inflammation Working Group.31 We selected variants that were associated with circulating CRP levels at genome-wide significance (p<5×10−8) and clumped these variants to a linkage disequilibrium (LD) threshold of r2 < 0.1 according to the European reference panel of the 1000 Genomes project.35 We estimated the variance in CRP levels explained by each of the SNPs by calculating the R2,36 and the strength of the instruments by calculating the F-statistic.37

(A) Shown is a simplified scheme of IL-6 signaling, which is induced by binding of IL-6 to the soluble or the membrane-bound form of its receptor (IL-6R). IL-6 signaling results in increased C-reactive protein (CRP) and Fibrinogen (Fg) levels and is associated with a higher risk of cardiovascular disease. (B) Pharmacological inhibition of IL-6R leads to increases in the levels of upstream regulators (IL-6 and sIL-6R), and decreases in the levels of downstream effectors (CRP and Fg) of the IL-6 signaling pathway, but its effects on cardiovascular disease remain unknown. (C) In the current MR approach, we selected genetic variants within the IL6R locus, which significantly associated with lower CRP levels, as instruments (proxies) for a downregulated IL-6 signaling, and explored their effects on ischemic stroke, coronary artery disease and other cardiovascular disease phenotypes.
*IL-6 signaling was determined by the effects of the instruments on CRP levels. The instruments were further validated by exploring their effects on other upstream regulators (IL-6, sIL-6-R) and downstream effectors (Fg) of IL-6 signaling.
sIL-6R, soluble IL-6 receptor.
In sensitivity analyses, we restricted our selection of instruments to SNPs within the IL6R gene (GRCh37/hg19 coordinates: chr1:154,377,669-154,441,926), to avoid potential pleiotropic effects through genes neighboring IL6R and increase confidence in the effects of the instruments through IL-6 signaling. As the instruments used in the current setting were not identified based on established biological effects, but solely on the basis of their statistical associations with CRP levels, in an additional sensitivity analysis, we restricted our genetic instrument to a single SNP (rs2228145) within the IL6R gene with well-established biological effects leading to a downregulation of the IL-6 signaling.20, 34, 38, 39
To disentangle the effects of IL-6 signaling from the respective effects of CRP, we selected SNPs associated with CRP levels at genome-wide significance (p<5×10−8) throughout the genome and clumped them to r2 < 0.1. We then performed MR analyses using all these SNPs as instruments, and performed 10,000 permutations for each outcome using 7 randomly selected SNPs (the same number as those used as instruments for IL-6 signaling). We further performed MR analyses using SNPs at the CRP locus as instruments (within a region of 300 kB upstream or downstream to the CRP gene; GRCh37/hg19 coordinates: chr1: 159,382,079-159,984,379).
To validate the instruments, we explored their associations with circulating levels of IL-6 and soluble IL-6R, which have previously been reported to increase as a result of both pharmacological inhibition and genetic downregulation of IL-6 signaling.20 We further explored association with fibrinogen levels, which is a downstream effector of IL-6 signaling and decreases after its blockade.20 The effects of genetic variants on IL-6 levels were obtained from a GWAS of 8,293 healthy individuals of Finnish ancestry.40 For soluble IL-6R levels, we used the summary statistics from the INTERVAL study exploring the human plasma proteome,41 as made publicly available through the PhenoScanner database.42 For fibrinogen levels, we used GWAS data from the CHARGE Inflammation Working Group on 120,246 European individuals.43
Outcomes
The primary outcomes for this study were ischemic stroke and coronary artery disease. Genetic association estimates for ischemic stroke and coronary artery disease were derived from the MEGASTROKE32 and CARDIoGRAMplusC4D44 consortia, respectively. Specifically, for ischemic stroke we used the European sub-dataset of MEGASTROKE (34,217 cases and 404,630 controls) to avoid population stratification with the CRP GWAS dataset, which also included solely individuals of European ancestry.32 The CARDIoGRAMplusC4D refers to a GWAS of 60,801 cases with coronary artery disease and 123,504 controls, primarily (77%) of European ancestry.44 Definitions for major ischemic stroke subtypes in MEGASTROKE followed the Trial of Org 10172 in Acute Stroke Treatment (TOAST) criteria with the following samples for analysis: large artery stroke (4,373 cases), cardioembolic stroke (7,193 cases), and small vessel stroke (5,386 cases; 404,630 controls for all subtypes).45 We further extended our analyses to other cardiovascular outcomes including myocardial infarction, aortic aneurysm, carotid artery plaque, peripheral artery disease, heart failure, atrial fibrillation, venous thromboembolism, deep vein thrombosis, and pulmonary embolism. The data sources and the sample sizes for these studies are presented in Table 1. For aortic aneurysm, heart failure, peripheral artery disease, deep vein thrombosis, and pulmonary embolism we used data from the UK Biobank, as described in Supplementary Methods.
Mendelian Randomization analyses
After extracting the association estimates between the variants and the outcomes and harmonizing the direction of estimates by effect alleles, we computed MR estimates for each instrument with the Wald estimator and standard errors with the Delta method.46 We then pooled individual MR estimates using fixed-effects inverse-variance weighted (IVW) meta-analyses.47 To provide clinically relevant results, all effect estimates were scaled to the CRP-decreasing effect of tocilizumab (8 mg/kg), between 4 and 24 weeks after administration (a decrease of CRP levels by 67%), as determined by a meta-analysis of 4 clinical trials.20 For the main IVW analyses, we performed power calculations and estimated the minimum and maximum effects that we had 80% statistical power to detect.48
The IVW method was our primary MR analysis approach. Although the selection of instruments on a specific gene reduces the possibility of invalid variants,49 the derived estimates might still be biased in case of directional pleiotropy. Hence, we further applied sensitivity MR analyses that are more robust to the inclusion of pleiotropic variants: the weighted median estimator, the contamination mixture method, and the MR Pleiotropy Residual Sum and Outlier (MR-PRESSO). The weighted median estimator provides consistent estimates as long as at least half of the variants used in the MR analysis are valid.50 The contamination mixture method constructs a likelihood function of the individual estimates and under the assumption that the estimates of the valid instruments would follow a distribution centered around the causal effect and any invalid instruments would follow a distribution around zero, it calculates MR estimates that would maximize this likelihood.51 The contamination method assumes that only some of the genetic variants used are valid instruments and it has been found to perform better than other methods under the presence of invalid instruments.52 Finally, we applied MR-PRESSO, which regresses the SNP-outcome estimates against the SNP-exposure estimates to test, using residual errors, whether there are outlier SNPs. Outliers are detected by sequentially removing all genetic variants from the analyses and comparing the residual sum of squares as a global heterogeneity measure (p-value for detecting outliers <0.05).53 MR-PRESSO then removes the identified outliers and provides outlier-corrected MR estimates.53 MR-PRESSO, is outlier-robust, but still relies on the assumption that at least half of the variants are valid instruments.53
For the primary analyses (associations between downregulated IL-6 signaling and risk of ischemic stroke or coronary artery disease), we set a statistical significance threshold at a two-sided p-value of < 0.05. For ischemic stroke subtypes and for other cardiovascular outcomes, we corrected for multiple comparisons with the Bonferroni method. Thus, the statistical significance thresholds were set at p<0.05/3=0.017 for the 3 ischemic stroke subtypes, and at p<0.05/9=0.0055 for the 9 cardiovascular outcomes. Associations not reaching these thresholds, but showing p-values <0.05 were considered suggestive. All analyses were performed in R (v3.5.0; The R Foundation for Statistical Computing).
RESULTS
Identification and validation of genetic variants as proxies of downregulated IL-6 signaling
Using our pre-defined selection criteria, we identified 7 SNPs to serve as instruments for downregulated IL-6 signaling (Table 2). Three of these instruments were situated within the IL6R gene (Supplementary Figure 1). The F-statistics of the 7 SNPs ranged from 81 to 764 indicating a low probability of weak instrument bias.37 Power calculations indicated that these instruments provide adequate statistical power (>80%) to detect ORs at the magnitude of 0.90 or lower for ischemic stroke and coronary artery disease regarding the effect of genetically downregulated IL-6 signaling (scaled to the CRP-decreasing effect of tocilizumab) (Table S1). We were further sufficiently powered (>80%) to detect ORs at the magnitude of 0.80 or lower for ischemic stroke subtypes.
The betas, standard errors, and p-values refer to associations of these SNPs with CRP levels.
To validate the 7 instruments, we explored associations of genetically downregulated IL-6 signaling with circulating IL-6, soluble IL-6R, and fibrinogen levels. In accordance with randomized clinical trials testing the effects of tocilizumab versus placebo (8 mg/kg),20 genetically downregulated IL-6 signaling was associated with higher circulating IL-6 and soluble IL-6R levels and with lower circulating concentration of fibrinogen with the strongest effects seen for soluble IL-6R levels (Figure 2).

(A) Effects of pharmacological inhibition of IL-6R on IL-6, sIL-6R, and Fg levels by administration of tocilizumab (8 mg/kg), as compared to placebo in a meta-analysis of 4 randomized clinical trials (RCT). Effects represent the standardized mean differences (SMD) in IL-6, sIL-6R, and Fg levels between 8 and 24 weeks after administration of tocilizumab (8 mg/kg), as compared to placebo. (B) Effects of genetic downregulation of IL-6 signaling on IL-6, sIL-6R, and Fg levels as determined by Mendelian Randomization (MR) analyses. Effects represent SMDs in IL-6, sIL-6R, and Fg levels.
*The SMDs for RCTs are derived from a meta-analysis of 4 studies.20
** The SMDs for the MR analyses are scaled to the CRP-decreasing effects of tocilizumab (8 mg/kg).
Genetically downregulated IL-6 signaling, ischemic stroke and coronary artery disease
We next explored associations between genetically downregulated IL-6 signaling (scaled to the CRP-decreasing effect of tocilizumab) with the risk of ischemic stroke and coronary artery disease (Figure 3). In the primary IVW analysis, downregulated IL-6 signaling was associated with a lower risk of both ischemic stroke (OR: 0.89, 95%CI: 0.82-0.97, p=3⨯10−3) and coronary artery disease (OR: 0.84, 95%CI: 0.77-0.90, p=7⨯10−6). The alternative MR approaches (weighted median, contamination mixture, MR-PRESSO) all showed consistent association estimates (Figure S2).

(A) Genetically downregulated IL-6 signaling in association with ischemic stroke and coronary artery disease as derived from IVW MR analyses either using the full set of genetic instruments (7 SNPs), or the restricted set of instruments (3 SNPs located within the IL6R gene). (B) SNP-specific effects regarding the associations of genetically downregulated IL-6 signaling with ischemic stroke and results derived from the IVW MR analysis. (C) Distributions of the effects of 7 randomly selected CRP-decreasing SNPs on risk of ischemic stroke and the position of the IL-6 signaling downregulating effect (7 SNPs included in our analyses).
*Odds Ratios for genetically downregulated IL-6 signaling are scaled to the CRP-decreasing effects of tocilizumab (8 mg/kg).
In sensitivity analyses restricted to the 3 instruments within the IL6R gene, we likewise found genetically downregulated IL-6 signaling to be associated with a lower risk of ischemic stroke and coronary artery disease (Figure 3 and Figure S2). Further restricting the analysis to a single SNP (rs2228145) with a well-described effect proxying the effects of pharmacological IL-6 signaling inhibition,20 we found presence of the allele linked to downregulated IL-6 signaling to be associated with lower risk of both ischemic stroke (OR: 0.88, 95%CI: 0.79-0.99, p=0.033) and coronary artery disease (OR: 0.75, 95%CI: 0.67-0.85, p=2⨯10−7).
To disentangle the effect of downregulated IL-6 signaling from the effect of CRP, we next performed MR analyses to explore associations between SNPs associated with CRP, and risk of ischemic stroke and coronary artery disease. These analyses showed no associations between genetically determined CRP levels and risk of either ischemic stroke or coronary artery disease independent of whether we used all variants reaching genome-wide significance (p<5⨯10−8) for association with CRP (187 SNPs), or whether we restricted the analyses to significant SNPs at the CRP locus (24 SNPs) (Figure S3). We further performed 10,000 permutations of MR analyses randomly selecting 7 out of the 187 SNPs associated with CRP. The effects of the 7 SNPs selected as instruments for downregulated IL-6 signaling on ischemic stroke and coronary artery disease were located at the 4th and 1st lowest percentiles of the respective distributions, corresponding to p-values of 0.04 and 0.01, respectively (Figure 3C and Figure S4), thus indicating that the effects of IL-6 signaling are independent of the effects of CRP itself.
Genetically downregulated IL-6 signaling and ischemic stroke subtypes
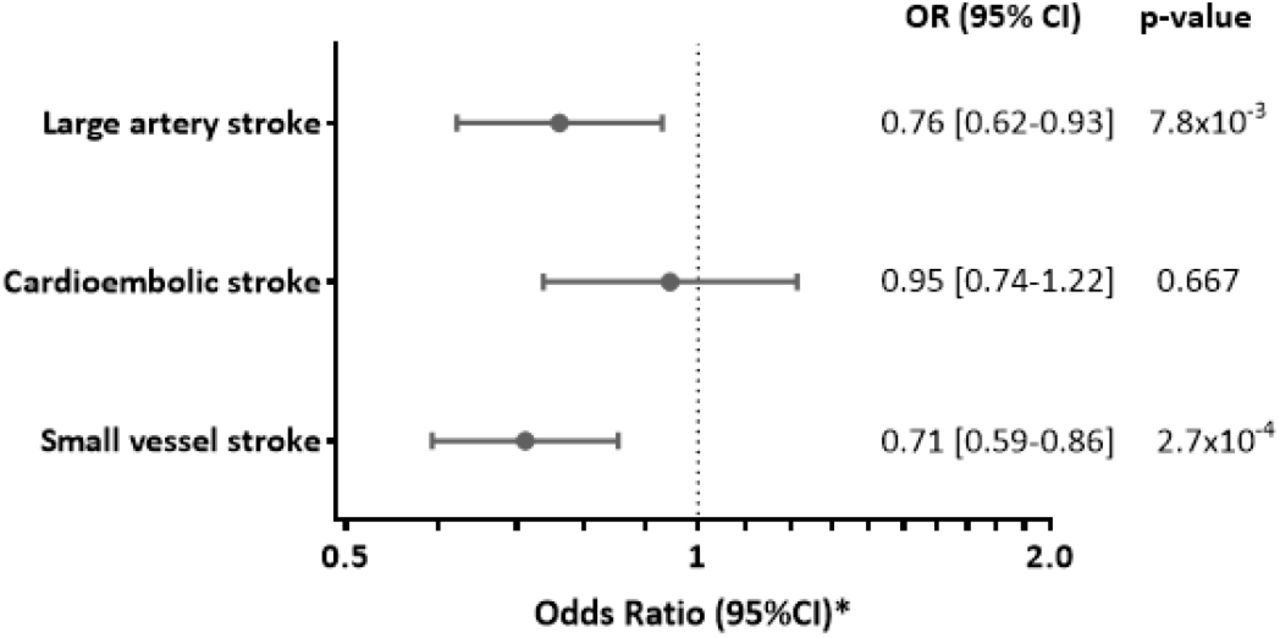
Focusing on etiological stroke subtypes (Figure 4), we found genetic downregulation of IL-6 signaling to be associated with a lower risk of large artery stroke (OR: 0.76, 95%CI: 0.62-0.93, p=8⨯10−3) and small vessel stroke (OR: 0.71, 95%CI: 0.59-0.86, p=3⨯10−4), but not cardioembolic stroke (OR: 0.95, 95%CI: 0.74-1.22, p=0.667). The results were stable in all MR sensitivity analyses, including when restricting the analyses to the instruments within the IL6R gene (Figure S5). We further found no associations between genetically determined CRP levels, as determined by SNPs throughout the genome or SNPs at the CRP locus, and any of the ischemic stroke subtypes (Figure S6). Similarly, in permutations of analyses including 7 randomly allocated SNPs throughout the genome, the effects of the SNPs proxying the downregulated IL-6 signaling on large artery and small vessel stroke, were at the 3rd and 0.1th percentiles (corresponding to p-values of 0.03 and 0.001), respectively, thus supporting that the observed effects were again independent of CRP (Figure S7).

The effects represent Odds Ratios (OR) derived from inverse-variance-weighted MR analyses.
*Odds Ratios for genetically downregulated IL-6 signaling are scaled to the CRP-decreasing effects of tocilizumab (8 mg/kg).
Genetically downregulated IL-6 signaling and other cardiovascular outcomes
In a last step, we expanded the analyses to other cardiovascular outcomes (Figure 5). Genetic predisposition to downregulated IL-6 signaling was associated with lower risks of myocardial infarction (OR: 0.88, 95%CI: 0.81-0.96, p=3⨯10−3) and aortic aneurysm (OR: 0.51, 95%CI: 0.37-0.68, p=1⨯10−5). We further found suggestive associations (p<0.05) with atrial fibrillation (OR: 0.83, 95%CI: 0.71-0.96, p=0.013) and carotid plaque (OR: 0.87, 95%CI: 0.77-0.99, p=0.041). In contrast, we found no significant associations with peripheral artery disease (OR: 0.91, 95%CI: 0.74-1.11, p=0.349), heart failure (OR: 0.90, 95%CI: 0.79-1.04, p=0.156), venous thromboembolism (OR: 0.98, 95%CI: 0.81-1.15, p=0.809), deep vein thrombosis (OR: 1.15, 95%CI: 0.94-1.40, p=0.183), and pulmonary embolism (OR: 0.92, 95%CI: 0.78-1.10, p=0.373).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effects represent Odds Ratios (OR) derived from inverse-variance-weighted MR analyses.
*Odds Ratios for genetically downregulated IL-6 signaling are scaled to the CRP-decreasing effects of tocilizumab (8 mg/kg).
DISCUSSION
Leveraging large-scale genetic data from multiple sources we identified variants serving as proxies for a genetic predisposition to downregulated IL-6 signaling and validated them using clinical trial data on pharmacological IL-6R inhibition. The identified proxies showed significant associations with a lower risk of both ischemic stroke and coronary artery disease. Among ischemic stroke subtypes, genetic predisposition to downregulated IL-6 signaling was associated with lower risks of large artery and small vessel stroke, but not cardioembolic stroke. Proxies for IL-6 signaling inhibition further showed significant associations with myocardial infarction and aortic aneurysm, and suggestive associations with atrial fibrillation and carotid plaque.
The MR association between genetically downregulated IL-6 signaling and lower risk of large artery stroke extends previous clinical,17, 18, 21 genetic,19, 20 and experimental54, 55 data demonstrating a key role of IL-6 signaling in atherosclerosis. By binding to IL-6R, IL-6 promotes downstream effects that include induction of macrophage recruitment56 and arterial smooth muscle cell proliferation,55, 57 and have been linked with plaque initiation,58 plaque destabilization,54 microvascular flow dysfunction,59 and adverse outcomes in the setting of acute ischemia.60 Moreover, pharmacological inhibition of IL-6R has been shown to attenuate atherosclerotic lesions in an experimental model of atherosclerosis.61 Our finding of an effect of genetic predisposition to downregulated IL-6 signaling on multiple atherosclerotic phenotypes (large artery stroke, coronary artery disease, myocardial infarction, aortic aneurysm, atrial fibrillation, carotid plaque) provides further support that IL-6 signaling is critically implicated in atherogenesis and atheroprogression and might represent a valid therapeutic target.
Notably, we found genetically downregulated IL-6 signaling to be further associated with small vessel stroke. There is only limited evidence regarding a role of inflammation in general and of IL-6 signaling in particular in cerebral small vessel disease.62 In a small prospective study of 123 patients with manifestations of cerebral small vessel disease, IL-6 circulating levels were associated with a higher risk of incident lacunes, a marker of small vessel disease on brain magnetic resonance imaging.63 However, cross-sectional analyses from larger population-based studies showed inconsistent findings for lacunes, silent brain infarcts and other manifestations of small vessel disease.64-69 While the specific mechanisms underlying our MR results remain unknown, our findings suggest that inhibition of IL-6 signaling aside from being a candidate treatment for atherosclerosis might also lower the risk of small vessel stroke.
Our results strongly support the candidacy of IL-6 signaling as a target for vascular prevention over and beyond previous data. The CANTOS trial targeted IL-1β rather than IL-6R and thus provided only indirect evidence for a benefit of interfering with IL-6 signaling.11, 21 Interestingly, the study further showed that part of the residual vascular risk after IL-1β inhibition could be explained by IL-6 levels, thus providing evidence that direct IL-6 signaling inhibition might represent a more effective strategy.22 Also, CANTOS was based on a population of individuals with coronary artery disease and explored a combined vascular endpoint rather than offering information on individual cardiovascular outcomes. With respect to stroke, there was a 7% reduction in incident stroke events in the IL-1β arm, which however did not reach statistical significance, possibly because of insufficient power.8 Our MR results provide evidence for directionally consistent effects of IL-6 signaling in multiple cardiovascular outcomes. Thus, our findings offer a solid basis for future clinical trials exploring the benefit of pharmacological IL-6R inhibition for the range of phenotypes examined here.
Interestingly, we found a particularly strong effect of genetically downregulated IL-6 signaling on aortic aneurysm. A role of IL-6 signaling in the pathogenesis of aortic aneurysm has been previously demonstrated by genetic studies.38, 70, 71 IL-6 signaling might contribute to the formation of aortic aneurysms through mechanisms aside from atherosclerosis, thus explaining the large effect. For instance, IL-6 signaling is a key pathway in the pathogenesis of large vessel vasculitides,72 which are strongly associated with the formation of aortic aneurysms.16, 73, 74
Our analysis provides no evidence for an association of genetically downregulated IL-6 signaling with cardioembolic stroke. In conjunction with the lack of significant MR associations with thrombotic phenotypes (venous thromboembolism, deep vein thrombosis, pulmonary embolism), our results do not support a role of IL-6 signaling in promoting coagulation and thrombosis. Yet, in accord with previous observational studies,75-77 we found IL-6 signaling to show a suggestive association with atrial fibrillation, the primary cause of cardioembolism and a common complication of coronary artery disease.78, 79 Given the relatively small magnitude of this association, any effect of IL-6 signaling on risk of cardioembolic stroke through atrial fibrillation would be expected to be small.
Our study has several strengths. Utilizing the most recent genetic data on CRP levels, ischemic stroke, and other cardiovascular phenotypes, we were sufficiently powered to show significant associations between genetically downregulated IL-6 signaling and multiple outcomes of interest. Using CRP levels, as a proxy for downstream IL-6 signaling enabled us to scale the derived association estimates to the respective effects of tocilizumab, as recorded in previous clinical trials, thus providing clinically meaningful estimates that might be comparable to those obtained from future trials. We further validated the effects of the selected proxies on upstream regulators (IL-6 and soluble IL-6R) and downstream effectors (fibrinogen) of IL-6 signaling, which were consistent with the effects observed with pharmacological inhibition of IL-6R. Finally, we could disentangle the effect of IL-6 signaling from the direct effect of CRP by determining the effects of CRP levels on risk of the examined outcomes and performing permutations for the effects of randomly selected CRP-decreasing variants.
Our study also has limitations. First, to proxy IL-6 signaling we used CRP levels, which are a downstream effect of the classical membrane-bound IL-6R-mediated signaling in hepatocytes.80 However, IL-6 also acts on other tissues not expressing the membrane-bound IL-6R, by binding to its soluble form, which is known as trans-signaling.80 Thus, our results may be interpreted as an effect of downstream regulation of classical IL-6 signaling but not IL-6 trans-signaling. Second, by design, our MR study assessed the effects of lifetime downregulated IL-6 signaling, which might differ from a shorter pharmacological inhibition. Third, there might be unknown pleiotropic effects of the genetic proxies used as instruments in the current study that might bias the associations. Of note, however, the results were remarkably consistent in sensitivity MR methods that are more robust to the inclusion of pleiotropic variants. Finally, our results were mainly based on individuals of European origin, and might thus not apply to other ethnic groups.
In conclusion, this study provides evidence for a causal effect of IL-6 signaling on ischemic stroke, particularly large artery and small vessel stroke, as well as a range of cardiovascular phenotypes. IL-6R blockade might represent a valid therapeutic target for lowering cardiovascular risk and should thus be further investigated in clinical trials.
Data Availability
All analyses presented in the manuscript have been conducted with publically available data. The instruments extracted for IL6 signaling are available in Table 1, for other authors to replicate our findings and use them in future work.
Funding
M. Georgakis was funded by scholarships from the Onassis Foundation and the German Academic Exchange Service (DAAD). D. Gill is funded by the Wellcome Trust. This project has received funding from the European Union’s Horizon 2020 research and innovation programme (No 666881), SVDs@target (to M. Dichgans) and No 667375, CoSTREAM (to M. Dichgans); the DFG as part of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198) and the CRC 1123 (B3) (to M. Dichgans); the Corona Foundation (to M. Dichgans); the Fondation Leducq (Transatlantic Network of Excellence on Pathogenesis of Small Vessel Disease of the Brain, to M. Dichgans); the e:Med program (e:AtheroSysMed, to M. Dichgans) and the FP7/2007-2103 European Union project CVgenes@target (grant agreement number Health-F2-2013-601456, to M. Dichgans).
Disclosures
No conflicts of interest to disclose.
Acknowledgements
We thank the following consortia for making data publicly available: MEGASTROKE Consortium, CARDIoGRAMplusC4D Consortium, AFGen Consortium, the YFS/FINRISK studies, and the INTERVAL study. This research has been conducted using the UK Biobank Resource (UK Biobank application 2532, “UK Biobank stroke study: developing an in-depth understanding of the determinants of stroke and its subtypes”).
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.
- 66.
- 67.
- 68.
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.
- 82.
- 83.
- 84.



