
Abstract
Objectives To evaluate the association between smoking behavior and type 2 diabetes risk using genetic data.
Research Design and Methods We obtained summary data from genome-wide association studies (GWAS) related to smoking behavior (n=1.2 million people) and type 2 diabetes (n=898,130) in individuals of European ancestry. Mendelian randomization analysis was performed using these summary-level statistics. Specifically, we assessed if genetically determined smoking initiation, smoking cessation, and smoking heaviness (number of cigarettes per day) modulated susceptibility to type 2 diabetes.
Results We observed that for each 2-fold increased odds of initiating smoking behavior corresponded with a 21% increased susceptibility to type 2 diabetes (OR = 1.21, 95% CI: 1.18-1.24, P=1×10−12). Our effect was consistent across instrumental variable methodological approaches, and we did not observe evidence of systematic biases to our effect estimate. When analyzed using an analogous approach, we found that the effect of smoking initiation increasing diabetes risk was equivalent to the effect of smoking behavior on increase coronary artery disease, an established causal risk factor for cardiovascular disease.
Conclusions Genetic data supports the hypothesis that smoking initiation corresponds with increased risk for type 2 diabetes. These findings are consistent with previous clinical epidemiological association between smoking behavior and elevated type 2 diabetes risk. Individual-level MR studies are necessary to estimate mediators and confounders related to this association.
Introduction
Type 2 diabetes is a leading causes of death worldwide (1). Type 2 diabetes and related comorbidities are increasing global epidemics, expected to place ever larger demands on health care systems. As such, risk factors that cause type 2 diabetes that can be readily intervened upon are crucial for prevention to public health.
Recent clinical and epidemiologic studies have demonstrated association with smoking with increased diabetes risk (2–6), suggesting that smoking might be a modifiable risk factor for type 2 diabetes. Smoking can perturb glycemic regulation (5,7), and murine studies recently revealed a common biological pathway underlying nicotine addiction and type 2 diabetes (8). Although smoking is associated with perturbations in a number of cardiometabolic traits (9–11), to our knowledge a causal genetic association between smoking and type 2 diabetes in humans has not yet been established.
Recent collections of genetic data for smoking behavior and diabetes risk now provide a vehicle through which evidence of causality can be evaluated, using the framework of mendelian randomization (MR). Formally, MR uses genetic variants associated with an exposure of interest (here, smoking behavior) to create an instrumental variable to estimate a potential causal effect of genetically-determined exposure on an outcome (here, type 2 diabetes). Because alleles are randomly allocated at meiosis, and due to the fact that genotype precedes phenotype, this approach with care can address issues of confounding and reverse causality that prevent inference in prospective or cross-sectional cohort studies (12). Using the largest genome wide association studies (GWAS) for both smoking behavior (13) and type 2 diabetes (14) reported to date, we applied the framework of MR to demonstrate evidence supporting smoking initiation as a risk factor for increasing susceptibility to type 2 diabetes.
Research Design and Methods
Genome-wide summary statistics collection
We analyzed publicly available GWAS summary statistics for smoking initiation (n=1.2 million individuals), smoking cessation (n=547,219), and cigarettes per day (n=337,334) (13), as well as type 2 diabetes (n=898,130) (14), and coronary artery disease (CAD, n=∼546,000) (15). We used data from individuals of European ancestry only. All data sets were analyzed in genome build hg19.
Genetic variant selection related to smoking exposures
We created genetic instruments related to smoking traits, including ‘smoking initiation’, ‘smoking cessation’ and ‘cigarettes per day’ (smoking heaviness) (13). For each smoking trait, we used PLINK (16) to identify single nucleotide polymorphisms within independent linkage disequilibrium blocks (EUR r2<0.01) in 250kb regions. Analogous instrumental variable (IV) results were obtained different LD pruning thresholds (i.e., r2<0.1 and r2<0.001, data not shown). After harmonization, full IVs included 341 single nucleotide variants (SNPs) for smoking initiation (Supplemental Table 1), 18 SNPs for smoking cessation (Supplemental Table 2), and 42 SNPs for cigarettes per day (Supplemental Table 3) on type 2 diabetes. Analogous workflows generated IVs for smoking traits on CAD (n= 342 SNPs, Supplemental Table 4).
Genetic correlations between smoking traits, type 2 diabetes, and coronary artery disease based on LD Score Regression (20).
We used mRnd (http://cnsgenomics.com/shiny/mRnd/, (17)) to estimate the strength F-statistics of these IVs (Supplemental Table 5). Smoking initiation or cessation were input as binary exposure variables, and smoking heaviness as a continuous exposure variable. We calculated proportion of genetic inheritance explained in each exposure variable per Shim et. al. (18). F-statistics greater than 10 were presumed to be devoid of weak instrument bias.
Mendelian randomization and causal effect estimation
We performed two-sample MR (TwoSample MR package v0.4.25 (12)) using R (v3.6.1). We present causal estimates from inverse variance weighted (random effects model), weighted median, and MR Egger regression methods. We analyzed for pleiotropic bias using Egger regression intercepts, wherein significant non-zero intercepts can imply directional bias among IVs (19).
Genetic correlation estimates
Genetic correlations were estimated using Linkage Disequilibrium Score Regression (LDSC) (20). Summary statistics were munged and analyzed for each trait. Presented data reflecting genetic correlation estimates (i.e., rg values).
Statistical analysis
Effects from smoking exposure on type 2 diabetes were analyzed using inverse variance weighted, weighted median, and MR-Egger regression measures. Because Cochran’s Q test (included in the TwoSample MR package v0.4.25 (12)) found heterogeneity our IVs, we utilized the random-effect model when performing inverse-variance weighted MR. We also performed a sensitivity analysis using IV pruning via MR-PRESSO (21) for the single trait (smoking initiation). The resultant IV included 325 SNPs from an initial 341, but MR results were not qualitatively different than the full instrument (see Supplementary Material), and thus we focus our presentation on MR results using all SNPs. Statistical significance was defined as P < 0.05.
We converted causal effect estimates into odds ratios using previously described methods (22). By multiplying causal effect estimates by ln(2) and taking the exponentiated value (=exp^[ln(2)*effect]), we calculated the change in type 2 diabetes odds ratio that corresponded with a 2-fold change in smoking initiation or smoking cessation risk. For smoking heaviness, a continuous variable, we exponentiated the causal effect estimate (=exp^[effect]) to reach a value reflecting the change in odds ratio per unit increase in exposure.
Coding scripts and data sets
All relevant coding scripts and data sets can be found on Github (https://github.com/thomchr/SmkT2D). All data and coding scripts are also available upon request.
Results
Increased odds of smoking initiation elevated risk for type 2 diabetes
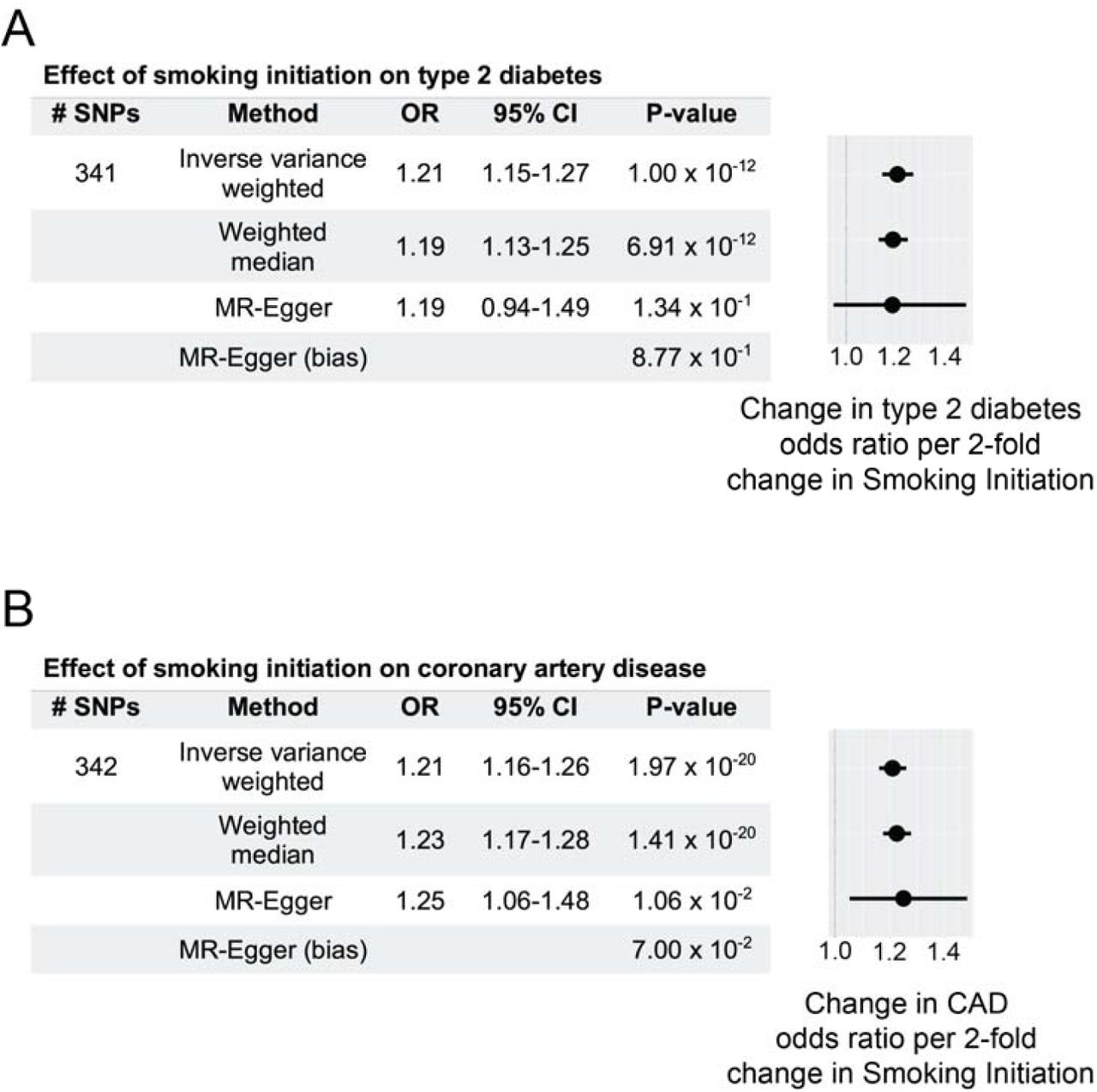
To estimate a causal effect between odds of smoking initiation and susceptibility to type 2 diabetes, we performed causal inference analysis using the framework of mendelian randomization. Using summary statistics obtained from genome-wide association studies of these traits, we generated an instrumental variable (IV) for smoking initiation (13), applied to data for type 2 diabetes (14). Our genetic instrument comprised 341 linkage-independent single nucleotide polymorphisms (SNPs, EUR r2<0.01) related to this smoking behavior. Our genetic instrument was not subject to weak-instrument bias (F-statistic = 4926). We observed that both inverse-variance weighted (P=1.0×10−12) and median-weighted (P=6.9×10−12) MR methods demonstrated positive association between genetically determined increase in odds of smoking initiation and increased susceptibility to type 2 diabetes (Fig. 1). As a sensitivity analysis, we performed the MR-Egger regression test, and found no positive evidence of systematic bias in our estimated effect (MR-Egger intercept term P=0.87). These data support the hypothesis that increased odds of smoking initiation corresponds with an increase of type 2 diabetes; each 2-fold increase in smoking initiation corresponding to a 21% increased type 2 diabetes risk (odds ratio, OR=1.21, 95% confidence interval = 1.18-1.24, by inverse variance weighted method).

A. Using a 341-SNP instrumental variable, two-sample MR shows that increased smoking initiation increases type 2 diabetes risk. Odds ratio (OR) estimates and 95% confidence intervals represent changes associated with 2-fold increase in smoking initiation exposure. MR-Egger intercept, a bias measurement, does not deviate significantly from zero. This validates effect (OR) estimates. Forest plot shows change in odds ratio for type 2 diabetes per 2-fold increased smoking initiation risk (mean ± standard error).
B. Using a 342-SNP instrumental variable, two-sample MR shows that increased smoking initiation increases coronary artery disease (CAD) risk. Odds ratio (OR) estimates and 95% confidence intervals represent changes associated with 2-fold increase in smoking initiation exposure. MR-Egger intercept, a bias measurement, does not deviate significantly from zero. This validates effect (OR) estimates. Forest plot shows change in odds ratio for CAD per 2-fold increased smoking initiation risk (mean ± standard error).
Increased risk of type 2 diabetes by smoking initiation is equivalent to that of coronary artery disease
To contrast our estimated effect to established causal relationships, we next performed an equivalent analysis using our smoking initiation instrumental variable on susceptibility to coronary artery disease (CAD). As with the above causal inference experiment with type 2 diabetes, our genetic instrument was devoid of weak instrument bias (F-statistic = 3780). We observed that a 2-fold increase in smoking initiation increased odds of coronary artery disease risk by 21% (OR = 1.21, 95% CI = 1.19-1.24 by inverse variance weighted method). The positive estimated causal effect on CAD agreed across MR methods (Fig. 1), and was consistent with previous reports (6). Strikingly, the estimated effect of smoking initiation on CAD risk was nearly identical to increased type 2 diabetes risk from this behavior (Fig. 1). These results indicate that the impact of smoking initiation on type 2 diabetes risk may have the same relative effect as this behavior on risk to CAD.
Smoking cessation and heaviness were not associated with type 2 diabetes risk
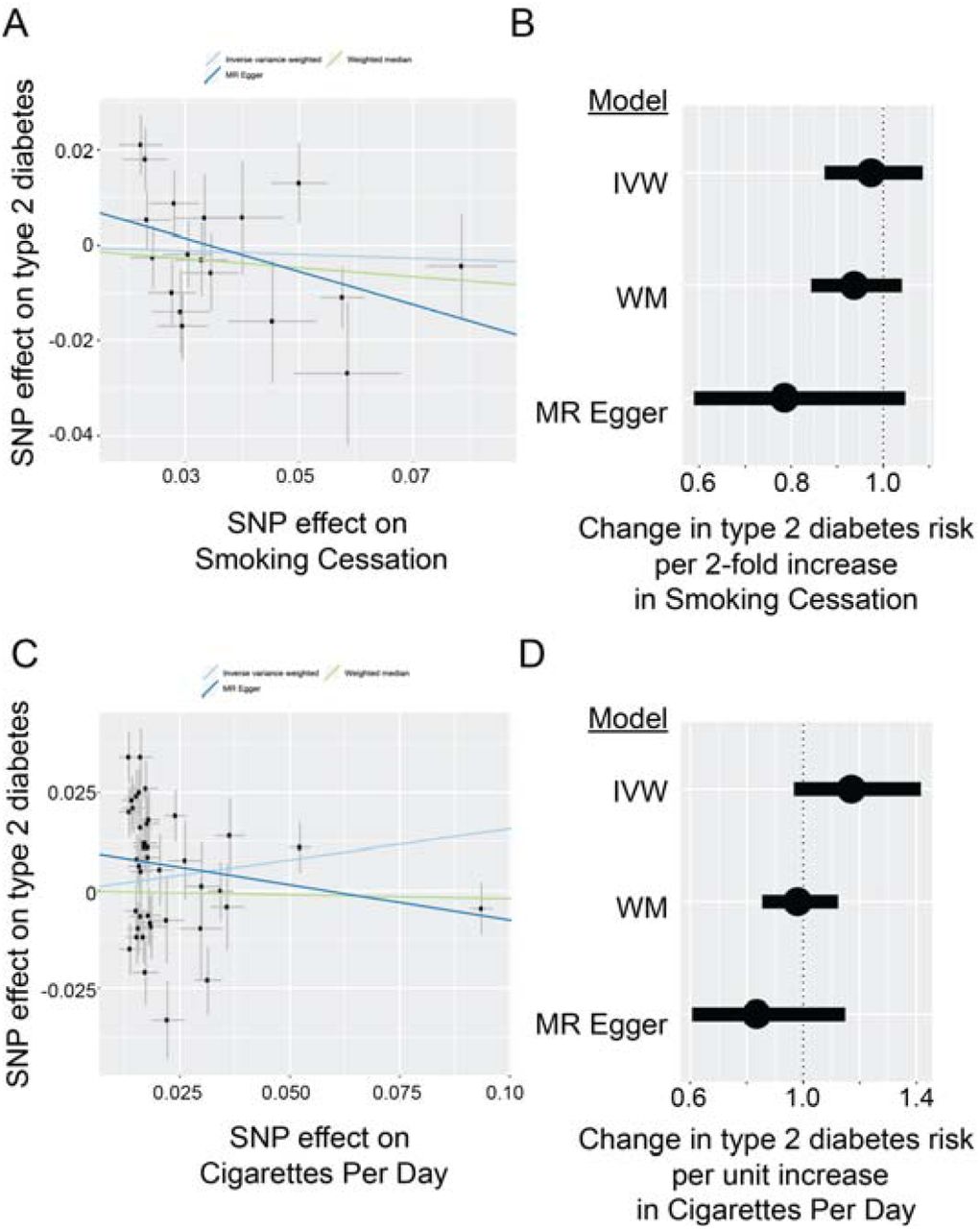
We next generated instrumental variables for additional smoking behaviors, including smoking cessation and smoking heaviness (cigarettes per day). The number of instruments for both of these traits were substantially smaller (n=18 and n=42 SNPs, respectively). F-statistics suggested that these instruments had no weak-instrument bias (F-statistic = 1737 for smoking cessation, F-statistic = 9897 for smoking heaviness). However, we did not observe evidence of association with either of these traits and type 2 diabetes risk (Supplemental Fig. 1 and Supplemental Table 6). A lack of association for these smoking traits with type 2 diabetes risk may be driven by lack of statistical power to detect relatively small effect sizes.
Smoking traits are genetically correlated with type 2 diabetes risk
If the results from the above were due to a lack of statistical power, we might expect to observe positive genetic correlation between smoking behaviors and type 2 diabetes risk. Therefore, we quantified the extent to which genetic susceptibility to these three smoking traits correlate with genetic susceptibility to type 2 diabetes. Using linkage disequilibrium score regression (LDSC) (20), we observed significant and positive genetic correlations between smoking traits and type 2 diabetes (Table 1).These results suggests that future genetic studies that explain smoking behavior and type 2 diabetes might clarify the effects of these smoking traits on type 2 diabetes.
Conclusions
Despite clinical observations (6), causal association between smoking and type 2 diabetes has not been established previously. Using the largest available repositories of meta-analyzed genetic summary statistics (13,14), we provide to our knowledge the first report of genetic evidence supporting increased risk of smoking initiation as a risk factor increased type 2 diabetes. The directionality of this finding is statistically and clinically significant, and concordant with the effects of smoking on CAD risk (Fig. 1). Although interpretation of these findings is limited to individuals of European ancestry, we expect that well-powered multi-ethnic GWAS will ultimately confirm this result in other populations.
These findings confirm and extend clinical observations relating smoking to increased type 2 diabetes risk, adding to numerous reports regarding negative effects of smoking on cardiometabolic health identified by medical practitioners and policymakers. A recent report detailed one mechanism linking smoking with type 2 diabetes risk (8). We anticipate that future validation research will establish clinically relevant biological mechanisms related to other loci as well. Although the effects of smoking may be mediated in part by comorbid cardiometabolic conditions, future multivariate MR studies will be necessary to investigate these associations. Importantly, these results show for the first time a genetic association between smoking and increased type 2 diabetes risk.
Data Availability
All data used for this work utilize summary data that is available in the public domain.
Supplemental Figure

{kind=link}
{kind=link}
A. Using an 18-SNP instrumental variable for smoking cessation, two-sample MR does not define an increased risk of type 2 diabetes. Scatter plot shows effect sizes (mean ± standard error) for smoking cessation and type 2 diabetes for each SNP in the instrumental variable, along with line of best fit for inverse variance weighted, weighted median, and MR Egger regression models.
B. Forest plot showing aggregated effects for smoking cessation on type 2 diabetes, shown as change in type 2 diabetes risk (odds ratio) per 2-fold change in smoking cessation “risk” (mean ± standard error) for inverse variance weighted (IVW), weighted median (WM), and MR Egger regression models.
C. Using a 42-SNP instrumental variable for smoking heaviness (number of cigarettes per day), two-sample MR does not define an increased risk of type 2 diabetes. Scatter plot shows effect sizes (mean ± standard error) for smoking heaviness and type 2 diabetes for each SNP in the instrumental variable, along with line of best fit for inverse variance weighted, weighted median, and MR Egger regression models.
D. Forest plot showing aggregated instrumental variable effects for smoking heaviness on type 2 diabetes, shown as change in type 2 diabetes risk (odds ratio) per unit increase in smoking heaviness (mean ± standard error) for inverse variance weighted (IVW), weighted median (WM), and MR Egger regression models.
Supplemental Tables
Instrumental variable data for MR experiments estimating effects of smoking initiation on type 2 diabetes. The rsid (hg19), chromosome, position, effect allele, non-effect allele, effect sizes and standard errors are shown for each SNP.
Instrumental variable data for MR experiments estimating effects of smoking cessation on type 2 diabetes. The rsid (hg19), chromosome, position, effect allele, non-effect allele, effect sizes and standard errors are shown for each SNP.
Instrumental variable data for MR experiments estimating effects of smoking heaviness (cigarettes per day) on type 2 diabetes. The rsid (hg19), chromosome, position, effect allele, non-effect allele, effect sizes and standard errors are shown for each SNP.
Instrumental variable data for MR experiments estimating effects of smoking initiation on coronary artery disease. The rsid (hg19), chromosome, position, effect allele, non-effect allele, effect sizes and standard errors are shown for each SNP.
Instrument strength F-statistics and related values from GWAS necessary to perform calculations (http://cnsgenomics.com/shiny/mRnd/, (17)). We calculated genetic risk explained in each exposure using a previously described method Shim et al (18).
Smoking cessation and smoking heaviness (cigarettes per day, Cig per day) do not reach statistically significant effects on type 2 diabetes risk based on inverse variance weighted (IVW), weighted median (WM) or MR Egger regression metrics. Odds ratio (OR) effects and 95% confidence intervals represent change in type 2 diabetes OR associated with 2-fold increased smoking cessation risk, or change in type 2 diabetes OR per unit increase in smoking heaviness (Cig per day). Significant Egger regression intercept values that deviate significantly from zero (e.g., Cig per day) invalidate effect estimates.
Acknowledgements
CST and BFV designed the study. CST, ZD, and BFV conducted, analyzed, and/or interpreted experimental data. CST and BFV wrote the paper. BFV acts as guarantor and corresponding author for this manuscript. The authors declare no conflicts of interest.
This work was supported by R56DK101478 and R01HG010067 (BFV), a Linda Pechenik Montague Investigator Award (BFV), T32HD043021 (CST), and an American Academy of Pediatrics Marshall Klaus Neonatal-Perinatal Research Award (CST). All authors confirm independence from funders and that all authors had full access to all of the statistics in the study. As such they can attest to the integrity of the data and the accuracy of the data analysis.



