
ABSTRACT
Germline pathogenic variants in TP53 are associated with Li-Fraumeni syndrome (LFS), an autosomal dominant cancer predisposition disorder associated with high risk of malignancy, including early onset breast cancers, sarcomas, adrenocortical carcinomas and brain tumors. Intense cancer surveillance for individuals with TP53 germline pathogenic variants has been shown to decrease mortality; therefore, accurate and consistent classification of variants across clinical and research laboratories is crucial to patient care. Here, we describe the work performed by the Clinical Genome Resource TP53 Variant Curation Expert Panel (ClinGen TP53 VCEP) focused on specifying the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines for germline variant classification to the TP53 gene. Specifications were applied to twenty ACMG/AMP criteria while nine were deemed not applicable. The original strength level for ten criteria was also adjusted due to current evidence. Use of the TP53-specific guidelines and sharing of clinical data amongst experts and clinical laboratories led to a decrease in variants of uncertain significance from 28% to 12% in comparison with the original guidelines. The ClinGen TP53 VCEP recommends the use of these TP53-specific ACMG/AMP guidelines as the standard strategy for TP53 germline variant classification.
INTRODUCTION
The TP53 gene encodes p53, a protein with essential roles in genome stability and key cellular functions such as cell cycle, metabolism, apoptosis, senescence and differentiation (Lane, 1992; Zerdoumi et al., 2017). Germline pathogenic variants in TP53 occur in ∼70% of individuals with Li-Fraumeni syndrome (LFS) (Schneider, Zelley, Nichols, & Garber, 1993), defined by patterns of personal and family history of certain early onset cancers, mainly pre-menopausal breast cancer, bone and soft tissue sarcomas, adrenocortical carcinomas and brain tumors. The cumulative incidence of cancer in LFS families is nearly 50% by age 31y for females and 46y for males, and up to 100% by age 70y for both (Mai et al., 2016). The extremely high penetrance has led to recommendations for intensive surveillance and other clinical management strategies (Ballinger et al., 2017; Evans, Birch, Ramsden, Sharif, & Baser, 2006; Kratz et al., 2017; Schon & Tischkowitz, 2018; Villani et al., 2016). Reported prevalence estimates range from 1:5,000 to 1:20,000 (Gonzalez, Noltner, et al., 2009; Lalloo et al., 2006), but recent studies suggest the frequency of germline pathogenic TP53 variants may be higher (Amendola et al., 2015; de Andrade et al., 2019; de Andrade et al., 2017).
Germline TP53 genetic testing was initially recommended for individuals meeting Classic LFS (Li et al., 1988) and/or Chompret criteria, most recently revised in 2015 criteria (Bougeard et al., 2015). Advances in next-generation sequencing have expanded the use of multigene panels as well as the inclusion of TP53 as a secondary finding for genome-scale sequencing (Kalia et al., 2017). This has led to an increased number of variants of uncertain significance identified in TP53, including in cancer patients who do not meet LFS criteria, and individuals without cancer (Bittar et al., 2019). Given the significant clinical and emotional challenges that come with an LFS diagnosis (Ballinger et al., 2017; Evans et al., 2006; Kratz et al., 2017; Schon & Tischkowitz, 2018; Villani et al., 2016; Young et al., 2019), it is essential to correctly assign TP53 germline variant pathogenicity.
The American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) variant curation guidelines are a series of generic criteria with different levels of strength for and against pathogenicity, incorporating evidence from multiple data sources(Richards et al., 2015). As part of the directive of the Clinical Genome consortium (ClinGen: https://clinicalgenome.org), specifications of these guidelines for specific gene/disease pairs are developed and documented by a Variant Curation Expert Panel (VCEP), and have been completed for cancer genes including CDH1 (Lee et al., 2018) and PTEN (Mester et al., 2018).
Herein, we present the scientific rationale and recommendations of the ClinGen TP53 VCEP to adapt the ACMG/AMP guidelines for the classification of TP53 germline variants and present results from pilot testing of the finalized guidelines.
METHODS
The TP53 VCEP followed ClinGen standard operating procedures (see https://clinicalgenome.org/curation-activities/variant-pathogenicity/documents/). The VCEP formed in 2015 by recruiting international TP53 experts knowledgeable in phenotype, molecular etiology, and functional processes. Twenty-four members contributed to at least one of three different evidence type working groups: Population/Computational, Functional, and Clinical. Each group reviewed the ACMG/AMP specifications in detail, incorporated this with critical review of the relevant literature and analysis of relevant data to inform evidence weights, and came to consensus for each specification.
VCEP members nominated variants for pilot testing the TP53-specific ACMG/AMP guidelines; 23 variants were selected to cover different molecular effects and the availability of data to allow assessing usability of different criteria. Seven variants from the International Agency on Research in Cancer (IARC) TP53 Database (Bouaoun et al., 2016) were included for their rich phenotypic information, but had no prior variant classifications. Thirteen variants from ClinVar database (Landrum et al., 2018) were used to balance the spectrum of suspected benign to pathogenic variants. Case level evidence available from clinical, research or clinical laboratory databases were provided, including information regarding cancer type(s) and family history, familial variant segregation, and de novo observations. Variant classifications (pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), likely benign (LB) and benign (B)) were also provided, which are referred to as prior expert assertions. Of note, variants pulled from ClinVar had assertions submitted by laboratories with representation on our VCEP, so that laboratory’s assertion in 2017 was considered as the prior expert assertion. Each variant was independently curated by two the five biocurators using the original ACMG/AMP guidelines and the TP53 specifications to test user interpretability. The criteria combinations for a given classification tier were as originally proposed (Richards et al., 2015), with the additional combination of very strong plus supporting criterion reaching LP for the TP53-specific guidelines supported by the Tavtigian et al. Bayesian rule combination calculator (Tavtigian et al., 2018). The resulting classifications were compared between biocurators, against prior assertions by the nominating expert(s) or contributing laboratories, and with ClinVar assertions (Landrum et al., 2018) when possible. During this phase, the TP53-specific guidelines were refined, and the final draft was shared with the ClinGen Sequence Variant Interpretation (SVI) Committee for approval.
RESULTS
TP53-specific variant curation criteria
Final TP53-specific ACMG/AMP guidelines are summarized in Table 1. Of the 28 original criteria, nine (PM3, PM4, PP2, PP4, PP5, BP1, BP3, BP5, and BP6) were excluded due to either limited data to support use of a rule code, irrelevance to TP53, or to avoid redundancy with another criterion specification. Additional details are shown in Supplementary Table S1. The remaining 19 criteria were modified by detailing the content and/or changing the strength level. Rationale for criteria specification are explained below.
Summary of the TP53-specificACMG/AMP criteria developed by the ClinGen TP53 Variant Curation Expert Panel
Population/Computational Working Group
Population data
BS1 and BA1 are criteria against pathogenicity based on the frequency of a germline variant in healthy individuals. To define the TP53 variant frequency cutoff for BS1, we calculated the maximum credible population allele frequency as reported by Whiffin et al. (Whiffin et al., 2017). Conservative values were used for prevalence and penetrance of TP53, which are 1 in 5,000 individuals (Lalloo et al., 2006) and 30% cancer risk similarly used by other hereditary cancer VCEPs (Lee et al., 2018; Mester et al., 2018). Genetic heterogeneity was set at 1 as TP53 is the only LFS-associated gene. This resulted in BS1 at 0.03%. BA1 was then defined as 0.1% based on a less conservative lifetime penetrance estimate of 70%, holding allelic and genetic heterogeneity at 1, and then increasing the derived maximum credible population allele frequency of 0.01% by on order of magnitude to arrive at a cutoff of 0.1%. For both criteria, a minimum of five alleles in a given population was required to ensure a comparable cohort. The TP53 VCEP specifically recommends the use of the most up-to-date non-cancer dataset of the gnomAD database (Karczewski et al., 2019), as this is the largest control database currently publicly available, and to ignore frequencies in Ashkenazi Jewish and Finnish populations due to founder effects (Kaariainen, Muilu, Perola, & Kristiansson, 2017; Shi et al., 2017).
The PM2 criterion uses absence in controls as evidence towards pathogenicity. Due to the overall rarity of TP53 germline variants (benign or pathogenic), the TP53 VCEP downgraded this criterion to supporting strength level.
PS4 requires case-control analyses, considered impractical for TP53 due to variant rarity and limited number of published studies. We instead developed a proband counting system to assign pathogenicity, based on the number of variant carriers meeting each of the existing clinical criteria for LFS. A likelihood ratio (LR) towards pathogenicity was calculated by dividing the proportion of carriers meeting classic LFS or Chompret 2015 criteria by the proportion of non-carriers meeting the same criteria, using data from individuals undergoing multigene panel testing at Ambry Genetics (Supplementary Figure S1). Results indicated that one proband with a variant meeting classic LFS or Chompret 2015 criteria would provide enough evidence to reach moderate (LR=15.57) or supporting (LR=3.37) strength level, respectively (Tavtigian et al., 2018). However, given the width of the confidence intervals, more conservative weights were assigned using a point system based on the number of probands meeting classic LFS or Chompret 2015 (Table 2). Additionally, it was decided that PS4 should not be applied if the variant also meets the population rules BS1 or BA1, to avoid coincidental accumulation of proband counts for common variants, following the approach previously developed for PTEN (Mester et al., 2018).
Point system created for the specifications of the PS4 rule*
The BS2 criterion uses presence of a variant in unaffected adults as evidence against pathogenicity. Two independent datasets (Ambry Genetics and GeneDx) were used to calculate how many observations of cancer-free adults at age 60 equated to strong evidence against pathogenicity. The LR towards pathogenicity was calculated by comparing the proportion of these individuals with a known TP53 pathogenic variant versus individuals without a TP53 pathogenic variant (Supplementary Table S2). The LRs estimated for observation of a variant in one healthy individual were 0.66 (Ambry dataset) and 0.28 (GeneDx dataset). Selecting the more conservative LR of 0.66, two to seven healthy individuals were considered necessary to apply BS2_Supporting (LRs ranging from 0.44 to 0.06), and eight or more to apply BS2 (LRs lower than 0.04) (Tavtigian et al., 2018). Given that the dataset used for this analysis included mostly female, and the associated risk of pre-menopausal breast cancer (Mai et al., 2016) for female TP53 carriers elevates their cancer risk compared with male TP53 carriers, it was stipulated that this criterion should be applied to female carriers only.
Computational and predictive data
PP3 and BP4 are commonly used criteria related to predictions using bioinformatic tools. Based on previous findings (Fortuno, James, Young, et al., 2018), an optimized version of Align-GVGD (Mathe et al., 2006) combined with BayesDel (Feng, 2017) (both included in the IARC TP53 Database R20 (Bouaoun et al., 2016)) were selected for bioinformatic prediction of TP53 missense variant pathogenicity. To assess prediction of effects on splicing, we selected MaxEntScan (Yeo & Burge, 2004) and Human Splicing Finder (HSF) (Desmet et al., 2009), commonly used and freely available tools that have shown good performance for predicting variant spliceogenicity for other cancer syndrome genes (Shamsani et al., 2019).
The BP7 criterion for silent variants was expanded to specify the use of MaxEntScan and HSF to predict any effects on splicing.
Strength level for PM5, which relies on previous observations of pathogenic variants at a given location as evidence of pathogenicity for other variants at the same location, was assessed as follows: The 493 non-functional variants from yeast transactivation assays(Kato et al., 2003) were assessed for the number of additional variants at the same amino acid residue that were also reported as non-functional, and compared to the number of functional or super trans variants (N=1239) at that position to generate a LR towards pathogenicity (Supplementary Table S3). Results indicated PM5 was applicable if two other pathogenic variants have been seen at a given amino acid residue (LR=6.46), but should be downgraded to supporting strength level if only one other pathogenic variant has been seen at the same residue (LR=2.90) (Tavtigian et al., 2018). Further, the following restrictions were added: (i) known P/LP variants must be based on classifications using the TP53-specific guidelines; (ii) variants using this rule must have equal or higher Grantham score (Grantham, 1974) than at least one pathogenic variant observed at that codon; (iii) this criterion cannot be used for any variant for which PM1 has been applied; (iv) splicing effects are excluded based on bioinformatic evidence from MaxEntScan and HSF.
PS1 may be applied as strong strength if effects on splicing due to the nucleotide change are excluded using splicing assay data, or downgraded to PS1_Moderate if only bioinformatic predictions (using MaxEntScan and HSF) are available as evidence against aberrant splicing. The comparative variant must have been classified as P/LP using the TP53-specific guidelines.
PVS1 is the only original criterion with a very strong strength level for pathogenicity, which applies to loss of function variants. The ClinGen SVI has published further guidance on this rule (Abou Tayoun et al., 2018), for which the TP53 VCEP has agreed to follow, including applying relative strengths for different types of null variants based on characteristics, such as variant type and location.
Functional Working Group
Functional data
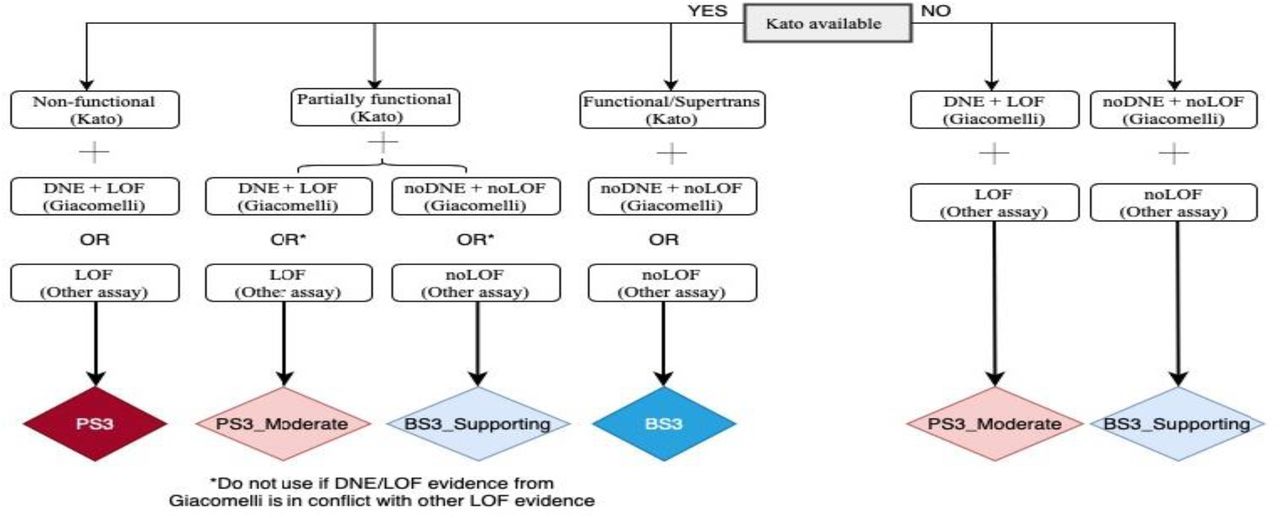
The functional-related PS3 and BS3 criteria, as originally described, do not specify required functional assays for a given gene. There are a number of published systematic functional studies of p53 missense variants, which measure transactivation activity(Kato et al., 2003), loss of function (LOF) (Giacomelli et al., 2018; Kotler et al., 2018) and dominant-negative effect (DNE) (Giacomelli et al., 2018). Matthews correlation coefficient was used to determine the relative performance of the different assay results for variant pathogenicity prediction; clinical reference sets were assumed pathogenic missense variants (present in classic LFS probands in the IARC TP53 Database R19 and absent in controls, N=52) and assumed benign missense variants (in controls from non-cancer gnomAD v2.1.1 or FLOSSIES (https://whi.color.com/) at a frequency higher than 0.0001 and not found in patients, N=31) (Supplementary Table 4). A decision tree considering availability and relative performance of the different assays is shown in Figure 3.
Hotspot data
The second part of the PM1 criterion related to protein regions was considered not applicable, as there is no known functional domain without benign variation in TP53. However, there are several well-described hotspots in TP53, occurring at amino acid positions 175, 245, 248, 249, 273, 282 (Brosh & Rotter, 2009; Fortuno, Pesaran, et al., 2019; Freed-Pastor & Prives, 2012), for which PM1 is applicable. Additionally, analysis of tumor DNA sequencing has identified a large number of TP53 variants as somatic hotspots, with information available at cancerhotspots.org (Chang et al., 2016; Chang et al., 2018). Following recommendations from the ClinGen Germline/Somatic Variant Curation Subcommittee (Walsh et al., 2018), it was specified that the PM1 criterion can also apply to somatically detected hotspots with ≥10 occurrences in cancerhotspots.org. This information is annotated in the IARC TP53 Database R20(Bouaoun et al., 2016).
Clinical Working Group
Segregation data
The original ACMG/AMP criteria use segregation as supporting strength for pathogenicity (PP1), allowing for stronger evidence if there is more segregation data, or a strong strength of evidence against pathogenicity when there is lack of segregation (BS4). Given the wide spectrum of cancer types that have been reported for TP53 carriers (Caron et al., 2016; Kratz et al., 2017; Olivier, Hollstein, & Hainaut, 2010), the criteria were specified based only on the number of meiosis of any LFS-associated cancer type, and number of families reported. Based on the gradations created previously for PTEN, the resulting TP53 specifications for PP1 are detailed in Table 1.
For BS4, which uses lack of segregation as evidence against pathogenicity, we considered potential issues due to the high de novo rate reported for TP53(Gonzalez, Buzin, et al., 2009), and specified use under these two scenarios: the variant segregates to the side of the family that does not meet LFS criteria, or the variant is present in three or more living unaffected individuals (where at least two of three are female) above 55 years of age (age specification consistent with Chompret 2015 criteria).
De novo data
The TP53 VCEP provides guidance for assigning the strength for the original de novo PS2 and PM6 criteria that should be based on the type of cancer and its relevance to the TP53 spectrum. This was accomplished using a point system incorporating parentage and proband cancer type which also allows for combination of points if there are multiple de novo reports of the same variant (Table 3).
Point systems created for the specifications of the PS2 and PM6 criteria and TP53-associated cancers with different strength levels*
All variants were annotated in relation to the transcript NM_000546.5 and protein NP_000537.3.
Cis/trans testing data
The use of BP2, which uses observation with another pathogenic variant as evidence against pathogenicity, was specified as follows: variant is observed in trans with a TP53 pathogenic variant (phase confirmed), or there are three or more observations with a TP53 pathogenic variant when phase is unknown. In this scenario, the variant must be seen with at least two different TP53 pathogenic variants (as specified by the TP53 VCEP).
Testing of the TP53 specifications on a pilot set of variants
There were 43 variants used for pilot testing. Classifications were compared between biocurators using the original ACMG/AMP and the TP53-specific guidelines, the existing assertions in ClinVar, and prior assertions by experts (Table 4).
Intra-biocurator consistency of variant classification was high: 72.1% using the original ACMG/AMP guidelines and 81.4% using TP53-specific guidelines (Table 4). There were fewer VUS using TP53-specific guidelines compared with the original guidelines (5/43 (12%) vs 12/43 (28%)) (Figure 2). Of the 42 pilot TP53 variants recorded in ClinVar in September 2019, there were 16 (38%) clinically relevant discrepancies in ClinVar at that time (see Table 4). Using the TP53-specific guidelines and sharing clinical data amongst experts and clinical laboratories, the majority of VUS were downgraded to LB (13/16; 81%), two moved to LP (2/16; 12.5%), and one remained at VUS (1/16; 6%).

Non-functional (Kato) = median transactivation activity ≤20%; Partially functional (Kato) = median transactivation activity >20 and ≤75%; Functional/Supertrans (Kato) = median transactivation activity >75%; DNE+LOF (Giacomelli) = p53WTNutlin3 Z-score ≥ 0.61 and Etoposide Z-score ≤ -0.21 = noDNE+noLOF (Giacomelli) = p53WTNutlin3 Z-score < 0.61 and Etoposide Z-score >-0.21. Other assays are available in IARC TP53 Database or original publications, and include in vitro growth assays in H1299 human cells from Kotler et al., (2018) with RFS score ≥ -1.0 for LOF and RFS score < -1.0 for noLOF; or colony formation assays, growth suppression assays, apoptosis assays, tetramer assays, knock-in mouse models.*If a variant does not match any of the possibilities shown, it is considered to have “no evidence to review” and no functional criterion can be applied.
Abbreviations: DNE = Dominant-negative effect; LOF = Loss-of-function.

{kind=link}
{kind=link}
Abbreviations: B = Benign, LB = Likely benign, VUS = Variant of unknown significance, LP = Likely pathogenic, P = Pathogenic, Conflicting = Clinically relevant conflicting interpretations of pathogenicity, and NA = Not Available.
DISCUSSION
The task of the TP53 VCEP was to specify the ACMG/AMP guidelines to assist with the clinical classification of variants in the TP53 gene. There is a rapidly growing number of individuals without personal or family history consistent with LFS who have germline TP53 variants (Batalini et al., 2019; Fortuno, James, & Spurdle, 2018), and cancer surveillance for people with TP53 pathogenic germline variants is time intensive and emotionally stressful (Ballinger et al., 2017; Kratz et al., 2017; Pantaleao et al., 2019; Villani et al., 2016; Young et al., 2019). Therefore, robust TP53 variant classification is essential for optimal clinical care.
Our pilot study demonstrated that the TP53-specific guidelines decreased the number of VUS compared with the original guidelines and increased the number of variants classified as not clinically relevant. Our study also showed strong intra-biocurator consistency, suggesting that the criteria appear to be straightforward to interpret; this should allow for fewer conflicting variant calls between laboratories.
One of the benefits of curating TP53 was the wealth of existing knowledge including, data readily available in public databases. The IARC TP53 database is a rich source of data that was helpful in assessing pathogenicity codes, including PS1, PS2, PS4, PM5, PM6, and PP1. The tremendous amount of data on p53 mouse models searchable by knockout variant of interest was also useful for adapting functional rule codes. Additionally, the TP53 VCEP benefited from data sharing amongst participating researchers, clinicians, and clinical laboratories. VCEP members added information pertaining to additional probands to those identified in the literature, to increase the number of pathogenic codes applicable to certain variants and to shift classifications from VUS to LP. Laboratory members also shared data from their large hereditary cancer panels that were helpful in using the benign codes BS2 and BP2.
Consistent with the ClinGen VCEP process, all of the TP53 classified variants are present in the ClinVar database with a summary of the classification process. More details about the evidence used to classify the variant is available through a link to the public access ClinGen Evidence Repository (https://erepo.clinicalgenome.org/evrepo/). The TP53 VCEP will continue to meet monthly to curate additional variants and submit them to ClinVar. Variants classified as LP or VUS will be reviewed at least every two years in the event new evidence has emerged, while variants classified as LB will be reviewed when new evidence is available or when requested by the public via the ClinGen website. It is also anticipated that these specifications will be reviewed as needed, to incorporate additional considerations and evidence types to improve TP53 variant classification (Fortuno, Cipponi, et al., 2019; Fortuno et al., 2020). The most up to date version of the ClinGen TP53 VCEP specifications is available at https://clinicalgenome.org/affiliation/50013/.
Data Availability
Most of the data that support the findings of this study are openly available in the IARC TP53 Database at https://p53.iarc.fr (version R20, July 2019). Other data supporting the findings of this study are not publicly available due to private or ethical restrictions. The publicly available Clinical Genome Resource (ClinGen) Evidence Repository contains all of the curated evidence for the variants submitted to ClinVar (e.g., https://erepo.clinicalgenome.org/evrepo/ui/interpretation/7b4332f9-03a6-43f1-afde-508d91bd92d5).
FUNDING
This Expert Panel is funded by National Human Genome Research & National Cancer Institutes (1U41HG006834, 1U01HG007437, 1U01HG007436, HHSN261200800001E, U41HG009650). The work of C.F. was supported by a University of Queensland (UQ) International Scholarship from the UQ School of Medicine. The work of M.F., K.C.A., and S.A.S. was supported by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health. The work of A.B.S. was supported by Australian National Health and Medical Research funding (ID1061779, ID1161589). The work of D.G.E. was supported by the NIHR Manchester Biomedical Research Centre (IS-BRC-1215–20007). The work of T.P.S. was supported by NIH-NCI K08CA234394 and R01CA242218.
CONFLICTS OF INTEREST
The following authors have no conflicts of interest to disclose: K.L., M.O., K.C.A., S.B., A.K.M.F., M.F., B.A.S., L.W., L.Z. The following authors have made extensive contributions to the TP53 literature and have previously published assertions on TP53 variants: C.F., P.L.M., L.D.A., D.G.E., P.J., K.M., T.P.S., A.B.S., S.A.S. The following authors are an employee, trainee or consultant for a commercial laboratory that offers genetic testing for TP53: T.P., R.H., K.M., S.E.P. J.M. is an employee of GeneDx/BioReference Laboratories, Inc./OPKO Health and has a salary as the only disclosure. The PERCH software, for which B.J.F. is the inventor, has been non-exclusively licensed to Ambry Genetics Corporation for their clinical genetic testing services and research. B.J.F. also reports funding and sponsorship to his institution on his behalf from Pfizer Inc. and Regeneron Genetics Center LLC.
DATA AVAILABILITY STATEMENT
Most of the data that support the findings of this study are openly available in the IARC TP53 Database at https://p53.iarc.fr (version R20, July 2019). Other data supporting the findings of this study are not publicly available due to private or ethical restrictions. The publicly available Clinical Genome Resource (ClinGen) Evidence Repository contains all of the curated evidence for the variants submitted to ClinVar (e.g., https://erepo.clinicalgenome.org/evrepo/ui/interpretation/7b4332f9-03a6-43f1-afde-508d91bd92d5).
ACKNOWLEDGEMENTS
The ClinGen TP53 Variant Curation Expert Panel would like to thank the ClinGen PTEN Variant Curation Expert Panel and the Executive Committee of the Hereditary Cancer Clinical Domain Working Group for sharing their experience with developing criteria specifications. We would also like to thank Steven Harrison, Leslie Biesecker and the remainder of the ClinGen Sequence Variant Interpretation Working Group for their guidance. Finally, we wish to thank present and past members of the TP53 VCEP, Maria Isabel Achatz, Rebecca Bassett, Jeffrey Bissonnette, David Goldgar, Sharisse Jimenez, Chimene Kesserwan, Deb Ritter, Leighton Telling and Mackenzie Trapp.
REFERENCES



