
ABSTRACT
BACKGROUND Mortality remains very high and unpredictable in COVID-19, with intense public protection strategies tailored to preceived risk. Genomic studies are in process to identify differences in host susceptibility to infection by the SARS-CoV-2 virus. Open source research publications are accessible to pre-genotyped individuals. Males are more at risk from severe COVID-19.
METHODS To facilitate development of Clinical Genetics support to the public and healthcare professionals, genomic structure and variants in 213,158 exomes/genomes were integrated for ACE2 encoding the SARS-CoV-2 receptor. ACMG/AMP-based pathogenicity criteria were applied.
RESULTS Across the 19 ACE2 exons on the X chromosome, 9 of 3596 (0.25%) nucleotides were homozygous variant in females compared to 262/3596 (7.3%) hemizygous variant in males (p<0.0001). 90% of variants were very rare, although K26R affecting a SARS-CoV-2-interacting amino acid is present in ~1/239 people. Modelling the “COVID-resistant” state where pathogenic alleles would be beneficial, nine null alleles met PVS1. Thirty-seven variants met PM1 based on critical location +/-PP3 based on computational modelling. Modelling a “COVID-susceptible” state, 31 variants in four upstream open reading frames and 5’ untranslated regions could meet PM1, and may have differential effects if aminoglycoside antibiotics were prescribed for pneumonia and sepsis.
CONCLUSIONS Males are more likely to exhibit consequences from a single variant ACE2 allele. Differential allele frequencies in COVID-19 susceptible and resistant individuals are likely to emerge before variants meet ACMG/AMP criteria for actionable results in patients. Prioritising genomic regions for functional study and ACMG/AMP-structured approaches to research-based presentation of COVID-19 susceptibility variant results are encouraged.
INTRODUCTION
The human population is currently undergoing natural selection for genomic variants that were not deleterious prior to the current SARS-CoV-2 pandemic. While COVID-19 infection can result in mild sequelae, life-threatening disease phenotypes are unfortunately common [1-6], intensive care mortality rates are reported in the order of 50-60% in recent large European series [4,5], and in the first four months of 2020, the disease claimed more than 250,000 lives despite often draconian measures to reduce the rate of spread of infection [7]. The impacts on society and commerce are unprecedented in peacetime.
The spectrum of disease severity in COVID-19 strongly points towards differences in host genetic susceptibility, and genomic studies are pending [8].
Given the proportion of the population who have already undergone whole exome or whole genome sequencing for other reasons [9-11], as new variants related to COVID-19 are reported in the scientific literature, those individuals are likely to seek reassurance or other information about their personal genetic risk. The pattern from Mendelian disorders was of less stringent assignment of variant pathogenicity status in the early reports [12], and for these pre-genotyped individuals, there may not be time between the publication of the COVID-19 associated variants, and the usual rigorous oversight applied by the Clinical Genetics Community, as exemplified by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) Guidelines [13].
Since the obligate receptor for SARS-CoV-2 is already known [1], this gene provides an opportunity to examine potential genetic risk ahead of publication of the imminent research studies. For infection to occur, SARS-CoV-2 has to recognise the membrane-bound host angiotensin-converting enzyme 2 [1]. The normal function of ACE2 is as a protease that catalyses the degradation of angiotensin II, with usually very small quantities of circulating enzyme released following cleavage of transmembrane ACE2.[14] Angiotensin II degradation is a crucial regulatory step which has multiple and essential cardio-protective consequences through modification of the renin-angiotensin-aldosterone system (RAAS).[14] Recently, the crystal structure of SARS-CoV-2 virus interacting with ACE2 was solved to 2.5-Ǻ resolution [15]. The viral Spike (S) envelope protein embeds into the surface of the human host cell, and is cleaved to subunits S1 and S2 (‘primed’) by TMPRSS2, augmented by cathepsin B/ L [16]. This process enables S1 to participate in receptor recognition, and S2 in membrane fusion. As detailed by Wang and colleagues [15], the S1 C-terminal domain (CTD) interacts with a single membrane-bound molecule of host angiotensin-converting enzyme 2 (ACE2), resulting in internalisation of the complex by the cell. The viral-receptor engagement was shown to be dominated by a series of strong polar contacts formed between specific viral and ACE2 amino acids, resulting in a solid network of hydrogen-bond and salt bridge interactions at the interface [15]. With these insights, it becomes feasible to begin to address whether variation in ACE2 may provide a rational for differing genotypic susceptibility to SARS-CoV-2.
The most obvious initial question regards males. More severe disease in males has been reported since the Jan 20th 2020 report of Huang and colleagues in their 41 reported patients [17], and later, the prominent mortality paper from Zhou and colleagues [2]. Recently, both Italian [5] and UK [4] intensive care units reported striking male excesses in more than 9,000 patients admitted to adult intensive care units. We have shown that males are more likely to be admitted to hospital, more likely to be admitted to adult intensive care units, more likely to die, and moreover have more severe biomarkers at all ages [6]. Taken together, the data point to a significant excess of males being more severely affected by COVID-19.
In addition to understanding male susceptibility, a more general understanding of who may be more at risk from COVID-19, and which individuals are more resistant to severe SARS-CoV-2 infection, will also be important for public and personalised health strategies.
The goal of this study was to define human genetic variation in the ACE2 gene and the potential for variants to be assigned as risk or protective alleles with respect to infection by SARS-CoV-2, using the structured approaches recommended by ACMG/AHP.
METHODS
ACE2 Reference Sequence
The 19-Apr-2020 update to NM_021804, the 3596 human ACE2 transcript variant encoding the ACE2 protein, was downloaded from RefSeq [18] and cross mapped to the National Center for Biotechnology Information Genome Reference Consortium Human Builds GRCh37/hg19 [19] and GRCh38/hg38 [20], utilising the University of Santa Cruz (UCSC) Genome Browser resources [21,22]. The exon 1 and 2 sequences 5’ to the methionine start codon were translated in all reading frames using ExPASY Translate [23].
Definition of ACE2 nucleotides of specific relevance to SARS-CoV-2 infection
The nucleotides encoding the amino acids participating in hydrogen bonding with SARS-CoV-2 in the 3D crystal structure [15], and those in the immediately adjacent residues, were mapped to the primary amino acid and nucleotide sequences of NM_021804. These, and the NM_021804 annotations for nucleotides encoding the transmembrane domain and protease cleavage sites of ACE2, were mapped to the two human genome builds currently in use, GRCh37/hg19 and GRCh38/hg38.
Assessment of ACE2 human variation
Variants in all ACE2 exons and flanking intronic regions identified by versions 2 and 3 of the Genome Aggregation Consortium (gnomAD) were downloaded between the 4th of April and 5 th May 2020 [24]. These largely non-overlapping datasets of 213,158 exomes and genomes from 111,454 male and 101,704 female datasets had been mapped to GRCh37/hg19 [19] (version 2, 141,456 samples) and GRCh37/hg19 [20] (version 3, 71,702 genomes). The variants were used to align the two human genome builds across the ACE2 locus. In describing homo/hemizygosity rates in males and females, the minimum and maximum potential number of individuals was recorded. To annotate against the ACE2 exons, where variants were identified in both versions, the mean allele frequencies were calculated.
COVID Disease States and Variant Interpretation
In order to interpret the variants, we constructed simple hypothetical models considering the initial susceptibility to COVID-19, or resistance to COVID-19 infection, as separate single gene disorders. Variant alleles that increase expression of transmembrane ACE2 protein, or otherwise enhance the receptor’s capacity to enable internalisation of SARS-Co-V into the host cell, are predicted to enhance COVID 19 risk, while loss of function variants are predicted to lower COVID 19 risk [25].
ACMG/AMP standards [13] were the primary source for classifying evidence for ‘pathogenicity’, modelling COVID resistance and COVID susceptibility potentially encoded by the ACE2 gene. These guidelines describe a structured approach to integrating evidence in favour of a functional consequence/pathogenicity, or alternatively benign status with likely or proven absence of impact on the phenotype under study. Multiple separate criteria are evaluated for a particularly gene variant based on allele frequency, functional consequence predictions, patient phenotype information, segregation studies in families, and other data from reputable sources [13]. The criteria span very strong [PVS]; strong [PS]; moderate [PM] and supporting [PP] evidence for pathogenicity; standalone [BA], strong [BS] and supporting [BP] evidence criteria for benign impact [13].
Variants were defined as meeting the ACMG/AMP criterion PVS1 for pathogenic/functional impact if they clearly resulted in loss of function, null alleles, i.e. multi-exon, pan exon, and frameshift insertions/deletions resulting in transcript ablation; start codon loss; stop codon gain; or disruption of ±2 splice site consensus sequences [13]. Other variants were evaluated in the context of predicted ACE2 critical residues, and evidence with respect to SARS-CoV-2 binding to date [15, 26]. Synonymous variants were recorded for allele frequency analyses but as none were in splice regions, were not considered further in the ACMG/AMP assignments.
Data Analysis
Aligned data were uploaded to STATA IC 15.1 (Statacorp, Texas, US) in order to generate descriptive statistics, to perform two way comparisons using χ2 and Mann Whitney tests, and to generate graphs.
RESULTS
ACE2 structure
As illustrated in Figure 1 and on all genome browsers, the human ACE2 gene comprises 19 exons which span 41kb of the X chromosome: the exact coordinates are chrX:15561033-15602158 on GRCh38/hg38, and chrX: 15579155-15620281 on GRCh37/hg19. The protein has a single membrane spanning domain encoded by exons 18 and 19, and a complex, hinge-bending extracellular structure [27]. The 24 key amino acids for viral-receptor engagement [15] are encoded by nucleotides in ACE2 exons 2, 3, 9 and 10. There are two membrane-proximal regions that are sites of cleavage to release circulatory ACE2. The first, encoded by exon 16, is recognised by ADAM-17, a disintegrin and metalloproteinase domain protein. The second, encoded by exons 17 and 18, is recognised by transmembrane protease, serine 2 TMPRSS2 (which also cleaves the viral Spike protein to subunits S1 and S2 [16]) and by transmembrane protease, serine 11D (Figure 1).

The 19 exons of the main transcript of ACE2 (NM_021804), sited on the X chromosome, and drawn as boxes to scale, with stylised untranslated regions (pale green), intervening introns and splicing events. Black numbers indicate exon first nucleotides, with the ATG start codon and TGA natural stop codon highlighted. Blue text indicates nucleotides encoding the signal peptide (SP), asparagines that are N-glycosylated (G), the transmembrane domain (TM), and two essential regions for cleavage by ADAM-17, and bytransmembrane proteases, serine 2 (TMPRSS2) and serine 11D (TMPRSS11D). Below the exons, amino acids (αα) numbered in blue refer to host ACE2 domains, with amino acids that hydrogen (H) -bond with the SARS-CoV-2 virus Spike protein CTD listed vertically, with precise position indicated at the point of the adjacent diamond. Above the exons, variants most likely to change COVID-19 susceptibility are described by coding nucleotide number (c. from methionine start i.e. +307 compared to exon 1 nucleotide 1), consequence (p. protein sequence; *, stop gain; fs, frameshift), and rs designation if available. Text is coloured as follows: Brown, loss of function alleles (start loss, stop gain, frameshift, and splice consensus variants); red, variants replacing amino acid residues directly hydrogen-bonding with SARS-CoV-2; purple, variants replacing an amino acid adjacent to a SARS-CoV-2 hydrogen bonding amino acid; green, variants affecting the cleavage sites, and black, variants affecting the transmembrane domain. Variants are also presented in Table 1.
Coding variants in ACE2
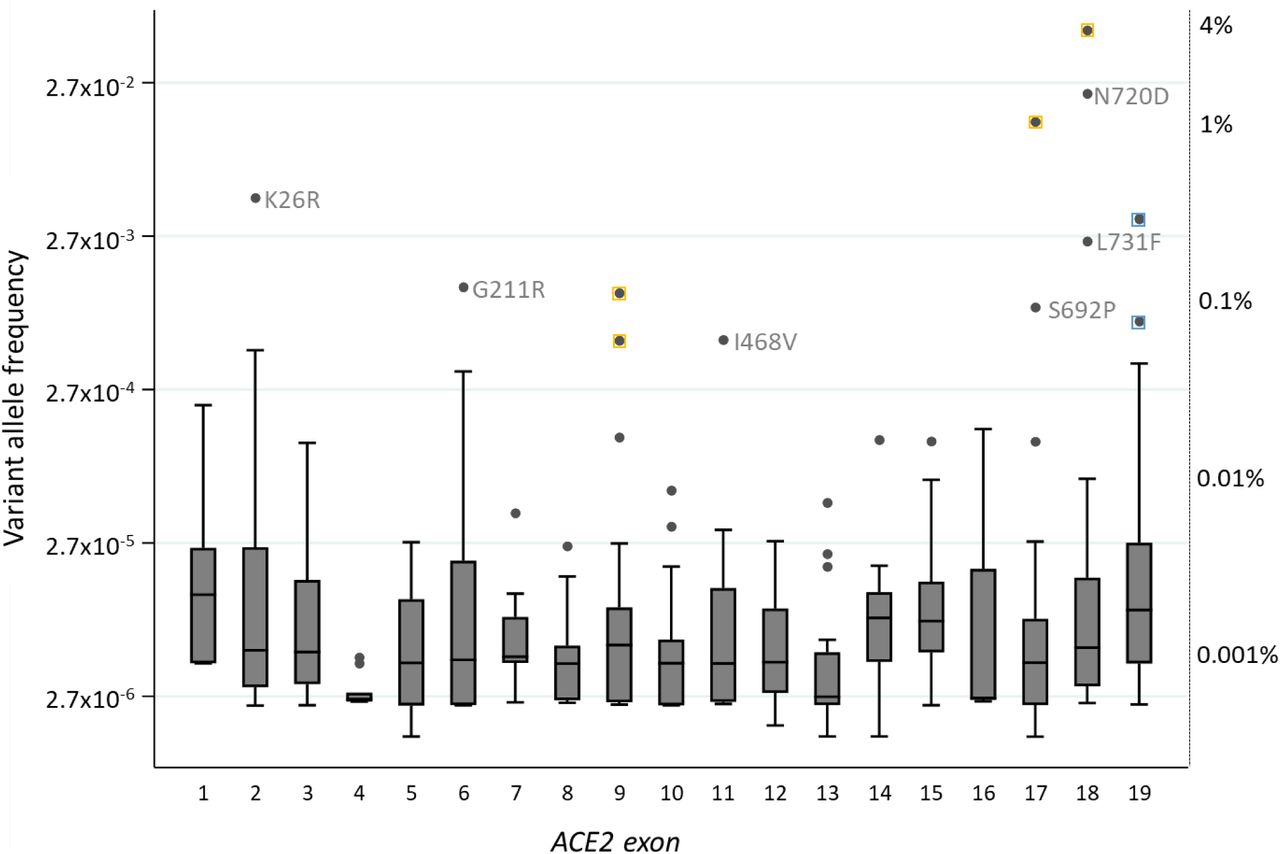
3,596 transcribed nucleotides are spliced to form the ACE2 mRNA (NM_021804). Across these and flanking splice site nucleotides, 483 variants were identified, with 305 (63.1%) present in only one of the two datasets. The variants included one whole gene duplication. The remainder were distributed across the ACE2 gene, and as shown in Figure 2, more than 90% were very rare, affecting fewer than 0.01% of the population. The twelve most common variants are annotated in Figure 2. Six of these resulted in amino acid substitutions, four were synonymous variants, and two were in the 3’ untranslated region of exon 19.

The allele frequency of all variants, grouped by exon and plotted on a logarithmic scale. Corresponding percentage frequencies are indicated on the right, anchored by key individual and median allele frequencies per exon. Box plots indicate median number and interquartile range, error bars 95 th percentiles, and dots are outliers. For the most common variants, yellow squares indicate synonymous variants where the nucleotide changes would not result in amino acid substitutions, and blue, non-coding variants in the 3’ untranslated region in exon 19. On the y axis, 2.7 represents e, the base of the natural logarithm that approximates to 2.71828.
Males are more likely to have an unopposed allele of ACE2
The implications of variant alleles differ for males with their single X chromosome, and females with two, due to Lyonisation when one female X chromosome is randomly inactivated in each cell [28]. Although approximately 1 in 5 x-chromosome genes can be transcribed from an inactive chromosome which would blur random distribution of DNA variant expression in females, ACE2 is not in this category [29]. Across the 19 exons of ACE2, only 9/3596 (0.25%) of nucleotides were homozygous for a variant allele in at least one female (Figure 3A) compared to 262/3596 (7.3%) present in a hemizygous state in males with their single X-chromosome (Figure 3B, χ2p<0.0001).

A) The number of females homozygous for (i.e. having two copies of) an ACE2 variant, plotted by the exon in which the nucleotide(s) are sited. B) The number of males hemizygous for ACE2 variants (i.e. having their only copy of the ACE2 sequence a variant) plotted by the exon in which the relevant nucleotide(s) are sited. Note the difference in y-axis scales between A) and B), similar patterns of distribution (reflecting overall variant allele frequency in the population, data not shown), and that most variation occurs in exons 17 and 18.
ACE2 exhibits few coding variants that generate an ACE2 loss of function allele
In gnomAD v2, the ACE2 gene exhibits substantial constraint against loss of function alleles [24]. In keeping with these metrics, across both version 2 and version 3 databases, there were only 9 clear loss of function variants: methionine start codon loss c.3G>A; stop codon gains c.347T>A, p.(Leu116*) and c.1967T>G, p.(Leu656*); splice donor losses c.802+1G>T, c.1442+2T>A, and c.1541+1G>T; splice acceptor loss c.584-5_584-2delAACA, and the frameshift variants c.1686dup, p.(Ser563Ilefs) and c.1265delG, p.(Gly422Valfs). These are illustrated by the brown text in Figure 1.
The nine loss of function variants were only found in the heterozygous state in females who had a second, normal (“wild-type”) allele on their other X chromosome.
In terms of COVID-19, all nine would meet the strongest ACMG PVS1 criterion in support of pathogenicity, noting that in this setting, this would be for the beneficial state of resistance to SARS-CoV-2 infection, since each would prevent any ACE2 / SARS-CoV-2 receptor production. Even so, applying ACMG [13], PVS1 alone is not sufficient evidence to classify as “pathogenic [beneficial in this setting]” or “likely pathogenic [beneficial in this setting]” as such assignment requires at least one further criterion of strong or moderate evidence.
ACE2 exhibits few coding variants that substitute amino acids that hydrogen bond with SARS-CoV-2
The ACE2 peptide sequence comprises 805 amino acids, 24 of which participate in hydrogen bonding with SARS-CoV-2. These amino acids are encoded by exons 2, 3, 9 and 10, as illustrated in Figure 1. The gnomAD databases indicated that 7 of the 24 SARS-Co-V-interacting amino acids are substituted by other amino acids in members of the human race, and a further 8 substitutions are reported affecting the immediate flanking amino acids. The population frequencies of these variant alleles ranged from variants affecting fewer than 1 in 100,000 people, to 0.4% (~1 in 239) for c.77A>G, p.(Lys26Arg/K26R) that substitutes the residue immediately preceding the SARS-CoV-2-bonding Thr27 and Phe28. In Figure 1, these variants are illustrated by red text if they replace an amino acid residue directly hydrogen-bonding with SARS-CoV-2, and purple if they replace an amino acid adjacent to a SARS-CoV-2 hydrogen bonding amino acid.
In contrast to the loss of function variants, across gnomAD versions 2 and 3, there was one female homozygote for c.77A>G, p.(Lys26Arg)/K26R, and according to the degree of overlap between the source DNAs, between 303 and 427 males who were hemizygous for c.52C>A, p.(Gln18Lys); c.55T>C, p.(Ser19Pro); c.76A>G, p.(Lys26Glu); c.77A>G, p.(Lys26Arg); c.103G>A, p.(Glu35Lys); c.109G>A, p.(Glu37Lys); c.246G>A, p.(Met82Ile); c.986A>G, p.(Glu329Gly); or c.1055G>T, p.(Gly352Val), as also listed in Table 1.
In terms of COVID-19, the 15 variant alleles for amino acids adjacent and directly interacting with SARS-CoV-2 could meet the ACMG/AHP moderate criterion of PM1 on the basis of affecting a critical domain. In this setting, this would more likely be for the beneficial state of resistance to SARS-CoV-2 infection, since the prediction would be of impaired ACE2/SARS-CoV-2 interaction, although the consequences of such binding impairment are not yet known. Even so, applying ACMG [13], PM1 alone is not sufficient evidence to classify as “likely pathogenic [beneficial in this setting]”. Such assignment requires at least two further moderate criteria, or a combination with up to 4 supporting criteria, only one of which can reflect allele frequency in disease and non-disease cohorts [13].
ACE2 exhibits few coding variants predicted to confer resistance to SARS-CoV-2 by substituting amino acids involved in maintenance of the transmembrane state
Thirty-one amino acids are essential for ACE2 stalk cleavage and for transmembrane domain (Figure 1). These amino acids are substituted in a number of rare variants.
Three missense variants were identified in nucleotides that encode the ADAM17 cleavage site, and are listed in Table 1. All had allele frequencies less than 0.000046. Thirteen missense variants were identified in nucleotides that encode the TMPR cleavage site (Table 1). For these, the respective allele frequencies were all less than 0.00026. Similarly for the transmembrane domain, the 6 identified missense variants (Table 1) had a maximal allele frequency less than 0.00011.
Across these 22 variants, no female was homozygote for any substitution. However, more than 50 males were hemizygous for at least one of seven variants in these regions (c.2086C>A, p.(Pro696Thr); c.2089A>G, p.(Arg697Gly); c.2115G>C, p.(Arg705Ser); c.2123G>A, p.(Arg708Gln); c.2122C>T, p.(Arg708Trp); c.2129G>A, p.(Arg710His); c.2128C>T, p.(Arg710Cys)).
For COVID-19, similar considerations apply as for the amino acids involved in direct hydrogen-bonding interactions with SARS-CoV-2: All 22 variant alleles could meet the ACMG moderate criterion of PM1 on the basis of affecting a critical domain. In this setting, variants that prevent membrane anchoring, or augment cleavage would be predicted to enhance resistance to SARS-CoV-2 infection, while those impeding cleavage of ACE2 would be predicted to enhance susceptibility. However, applying ACMG [13], PM1 alone is not sufficient evidence to classify as likely pathogenic, and allele frequency in disease and non-disease cohorts would only meet one of the two of four additional criteria required [13].
Upstream open reading frames in the ACE2 gene
Working from genomic coordinates, additional exonic ACE2 variants were retrieved in the first exon, and across all untranslated nucleotides of exon 19. The region spanning exons 1 and 2 is illustrated in Figure 4. On ExPASy translation, the first exon, together with the 5’ region of the second exon, contains 4 non-overlapping upstream open reading frames (uORFs) which are known in general to be able to stimulate or inhibit translation of main ORFs [30]. The first uORF is in-frame with the main transcript, the second, third and fourth are in the +1 reading frame. As indicated in Figure 4, 12 variants were in three of the uORFs, a further 7 variants in the uORF 5’untranslated regions, and 14 in the exon 2 5’UTR for the main transcript. All were rare, with allele frequencies less than 0.00028.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACE2 exon 1 and the start of exon 2 with upstream open reading frames (ORFs) to scale, stylised intervening intron/splicing event, and variants approximately positioned. Note that compared to Figure 1, the 5’ “untranslated” region has been deconstructed into open reading frames and 5’/intervening untranslated regions. Grey indicates the in-frame ORF, with variants listed in grey text. Lilac indicates alternate reading frame ORFs with variants listed in purple text. Pale green indicates the intervening untranslated regions, with the relevant variants listed in green text.
No variant was present in a homozygous state in females though 14 males were hemizygous for a variant in exon 1 (rs1049986543, rs954681758, rs934738609, rs1432973822, rs867425621), and 22 males hemizygous for a variant in the untranslated regions of exon 2 (rs981244191, rs1452729298, rs370596467, rs762634913, rs1388994308, rs1455256395).
DISCUSSION
We have shown that the ACE2 gene on the x-chromosome provides a biological rationale why males and females have different risk profiles for COVID-19 infection. Within coding nucleotides, identified variants were rare, and complete loss of function alleles only observed in the heterozygous state in females. For these, and alleles sited within critical domains defined by other methods, full ACMG criteria for “pathogenicity” would not be met easily, even if different patterns of distribution were captured in very large COVID-19 sequencing initiatives. The first and proximal second exons with multiple uORFs are identified as a source of further variation, though again each individual variant is rare.
The current manuscript utilised the gnomAD databases due to their very wide population coverage, ease of accessibility, and familiarity to Clinical Geneticists [24]. It is widely considered that variant databases are already saturated for common alleles although further rare variants are expected to emerge, particularly from previously under-represented populations [31]. Thus, although a limitation of the current study is that not every human genomic database was examined, notably, a wider number of coding ACE2 variants were retrieved compared to other publications [25], most likely due to the combined use of both the GRCh37/hg19-mapping and more recent GRCh37/hg19-mapping gnomAD databases as in [6]. By design, the study was limited to ACE2, in order to illustrate principles, and could be expanded to additional potential regions of genomic risk such as alleles affecting expression of TMPRSS2 or cathepsin B/ L [16].
A crucial question for the Clinical Genetics community is how strictly putative risk and protective alleles should be defined, particularly if they may become part of public health policies, as for underlying health conditions [32]. Given the individual rarity of coding variants illustrated by ACE2, a sufficient number of carriers may never be exposed to SARS-CoV-2 to establish whether or not they would be more or less infected/affected than expected. Systematic in vitro modelling of infection rates in ACE2 heterozygous cells may provide evidence that would meet the PS3 functional studies criterion, thereby enabling variant classification and advancing core knowledge about a protein whose molecular regulation has not been centre-stage in recent decades. Computational modelling for the small number of specific missense variants at amino acids directly interacting with SARS-Co-V has commenced, and provides an ACMG/AMP strong criterion of evidence (PS3) for a number of alleles: Hussain and colleagues demonstrated that two SARS-Co-V interactions of ACE lysine 353 are predicted to be absent if the apparently ‘distant’ serine 19 is substituted by proline (S19P) [26]. Modelling docking poses of SARS-Co-V-2 suggested intermolecular contacts were also fewer than predicted for E329G, and inter-residual interaction maps indicated that SARS-Co-V interaction with ACE Q42 was absent in E35D, E37K, M82I, and E329G. Additionally, a significant change in the estimated free energy of ACE2 for S19P and K26R implied these may adversely affect protein stability compared to wildtype [26]. Thus PM1 and PP3 (in terms of “pathogenicity” for a COVID-resistant state) would be met for these six variants, including ACE2 c.77A>G, p.(Lys26Arg) which is the second most common ACE2 missense substitution in the human population (Figure 2).
The consequences of an unopposed loss of function ACE2 allele in males are not yet known, but the paucity of loss of function alleles in this X-chromosome gene points towards evolutionary constraint based on the role of ACE2 as a critical homeostatic mediator through the renin angiotensin system [14]. The similarly low allele frequencies for missense substitutions in the SARS-Co-V binding region and the stalk cleavage sites, suggest these too have been under evolutionary constraint, in keeping with the essential functional catalytic role of ACE2 as a transmembrane carboxypeptidase. It therefore appears that if common resistance or susceptibility alleles to SARS-Co-V emerge, they are unlikely to be found in the ACE2 coding regions. Currently the Genotype-Tissue Expression (GTEx) project identifies 2,053 expression quantitative trait loci (eQTLs) for ACE2 based on mapping windows 1Mb up and downstream of the transcription start site [33]. While these loci will likely harbour SARS-CoV-2 genomic susceptibility and resistance alleles, and some are very common, with minor allele frequencies approaching 0.5, the significant linkage disequilibrium means selecting individual loci for functional evaluations may be challenging [34].
For immediate clinical considerations, regulation of the ACE2 uORFs is suggested as an initial focus for functional assays in the presence and absence of relevant variants. This is because aminoglycoside antibiotics (such as gentamicin, amikacin, neomycin, tobramycin and kanamycin) which are used in vitro to manipulate uORFs via ribosomal read-through of termination codons [35], are quite frequently used in intensive care settings to treat very sick patients including those with COVID-19. In vitro studies would provide a rationale for whether these agents have no effect, increase or decrease ACE2 transcription and thus whether no effect, beneficial or deleterious effects on disease progression would be predicted. Also of relevance would be to examine transcript responses to more commonly used pharmaceutical RAAS inhibitors such as angiotensin I converting enzyme inhibitors, angiotensin II type 1 receptor antagonists, and mineralocorticoid antagonists. These are used to treat hypertension [14], though more complex experimental design, potentially in vivo, would be required to test these agents in an integrated system. Such studies should also incorporate consideration of the alternate ACE2 transcripts that do not produce functional ACE2 protein [21,22,33].
In conclusion, given the obligate host receptor for SARS-CoV-2 is located on the X chromosome, simple genetic considerations supported by population-based genotyping provide a plausible explanation of why more men than women are severely affected by COVID-19. The data and concepts further emphasise the importance of increasing targeted and personalised care to males, and highlight priority regions of the human genome for interrogation. These focus not only on ACE2 amino acids interacting with SARS-CoV-2 and in critical sites that maintain ACE2 in its transmembrane form, but also the upstream reading frames where read-through may be modified by treatments used in late stage disease. Given the very large number of COVID-19 host genomics studies [8], differential allele frequencies will probably emerge before variants meet ACMG/AMP criteria for actionable results in patients. Stewardship of variant calls is likely to be required, and in the interim, ACMG/AMP-structured approaches to research-based presentation of COVID-19 susceptibility variant results is encouraged.
Data Availability
Data are available in the primary data sources (references 18-24). We are in the process of developing an accessible database that will be open access following peer review publication.
Data Availability Statement
Data are available in the primary datasources (references 18-24,33). We are in the process of developing an accessible database that will be open access following peer review publication.
Ethical Approvals
Ethical approvals were not required as all data reported were already anonymised and in the public domain.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Acknowledgements
We gratefully acknowledge the research laboratories and bioinformatics groups whose open source materials made these analyses feasible. The work received no specific funding. CLS acknowledges infrastructure support provided by the NIHR Imperial Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of funders, the NHS, the NIHR, or the Department of Health and Social Care.
References



