
Abstract
Background & aims Sex differences in non-alcoholic fatty liver disease (NAFLD) are well known and yet, most of the studies available in the literature do not include this factor when analysing the data. Here we present a functional meta-analysis of NAFLD studies to detect the molecular mechanisms involved in its progression, distinguishing any differences relative to patient sex.
Methods We systematically reviewed the Gene Expression Omnibus (GEO) database functional Gene Ontology (GO) terms detailed in transcriptomic studies, following the PRISMA statement guidelines. For each study, we compared the progression from steatosis (NAFL) to steatohepatitis (NASH) in premenopausal women and men using a dual strategy: a gene-set analysis and a pathway activity analysis. The functional results of all the studies were integrated in a meta-analysis used to detect functions that were differentially affected in these groups, according to these two methods.
Results A total of 105 abstracts were reviewed and 7 studies, which included 624 patients, were finally analysed. The meta-analyses highlighted significant functions in both sexes. In premenopausal women, the overrepresented functions referred to DNA regulation, vinculin binding, interleukin 2 (IL-2) responses, negative regulation of neuronal death, and the transport of ions and cations. In men, they referred to the negative regulation of IL-6 and the establishment of planar polarity involved in neural tube closure.
Conclusions Our meta-analysis of this transcriptomic data provides a powerful approach to identify sex differences in the progression from NAFL to NASH. We detected relevant biological functions and molecular terms that were affected differently between premenopausal women and men. Changes in the immune responsiveness between men and women with NAFLD suggested that women have a more immune tolerant milieu while men have an impaired liver regenerative response. These results open the door to more pathophysiological studies into the differential role of IL-2 and IL-6 in NAFLD, and eventually to personalised treatments for this disease.
Lay summary Progression from NAFL to NASH differently affects cellular functions in women and men. Here we systematically reviewed publicly available transcriptomic data and then performed a meta-analysis to find these affected functions. Thus, we identified 13 biological functions implicated in the progression of NAFLD that were differentially affected by sex.
Highlights
Transcriptomics meta-analysis detects significant sex differences in NAFLD progression.
Interleukins 2 and 6 are upregulated in premenopausal women compared to men.
Changes in the immune responsiveness may explain the sex-differences observed.
Sex differences need to be taken into account in the NAFLD studies.

Introduction
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of liver disorders ranging from fat accumulation in hepatocytes (NAFL) to non-alcoholic steatohepatitis (NASH), which can all potentially lead to cirrhosis or liver cancer. Interest in this disease has increased in recent years because of its worldwide impact on health, given that it is now the most common liver disease in developed countries [1]. It is estimated that the worldwide prevalence of this disease is around 25%, although it is more common in South America and the Middle East and less prevalent in Africa [2].
Sex also affects the distribution of this pathology, with current studies showing a higher prevalence in men and postmenopausal rather than premenopausal women [3]. Indeed, the liver shows the second largest amount of sexual dimorphism in humans. Both physiological and pathological hepatic processes, such as the detoxifying metabolism of cholesterol, as well as the prevalence of hepatic diseases differ between men and women [4]. Hence, the progression of NAFLD and the effect of treatments are also likely affected by patient sex [5]. Nonetheless, most studies on this disease do not consider metabolic differences between the sexes [6].
Although the presence of NAFLD can be strongly suspected based on imaging results, clinical analyses, and the presence of comorbidities, histological analysis is the only definitive way to diagnose it [7–9]. Because of its invasive nature, the fact that a liver biopsy is the only completely reliable technique to diagnose NAFLD is a limitation when conducting studies in human patients.
The progression of the disease is different in each patient, and can have a considerable impact on the clinical consequences to the individual. Patients in the initial stage of the disease (NAFL) have a low risk of adverse outcomes [10], but its progression to NASH increases the possibility of clinical complications, both liver-related and otherwise.
In this context, we carried out a systematic review of the literature related to this topic and selected transcriptomic studies which had deposited data in the Gene Expression Omnibus (GEO) datasets database [11]. We used these to clarify the molecular basis of the progression from NAFL to NASH in premenopausal women and in men. Sex differences related to this progression may have important clinical implications to patients, and could potentially favour a personalised approach to this disease in the future.
Materials and methods
Search studies
This review was conducted in August 2019, following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement guidelines [12]. We searched the GEO datasets database [11] using the keywords: “NAFLD”, “NAFL”, “steatosis”, “NASH”, and “steatohepatitis” for transcriptomic studies published in English.
Study exclusion criteria
We applied the following exclusion criteria: (i) studies conducted in organisms other than humans; (ii) studies without information about the sex of the participants or that had not included both sexes; (iii) studies without individuals from the NAFL and NASH stages of the disease; (iv) studies on methylation; (v) studies in which the disease had not been diagnosed with a biopsy. In the latter case, we decided to require the use of a biopsy as the method for NAFLD and NASH diagnosis because other less invasive methods such as conventional imaging techniques have a low sensitivity to detect mild steatosis and/or to differentiate NAFL from NASH [8].
Bioinformatics analysis strategy
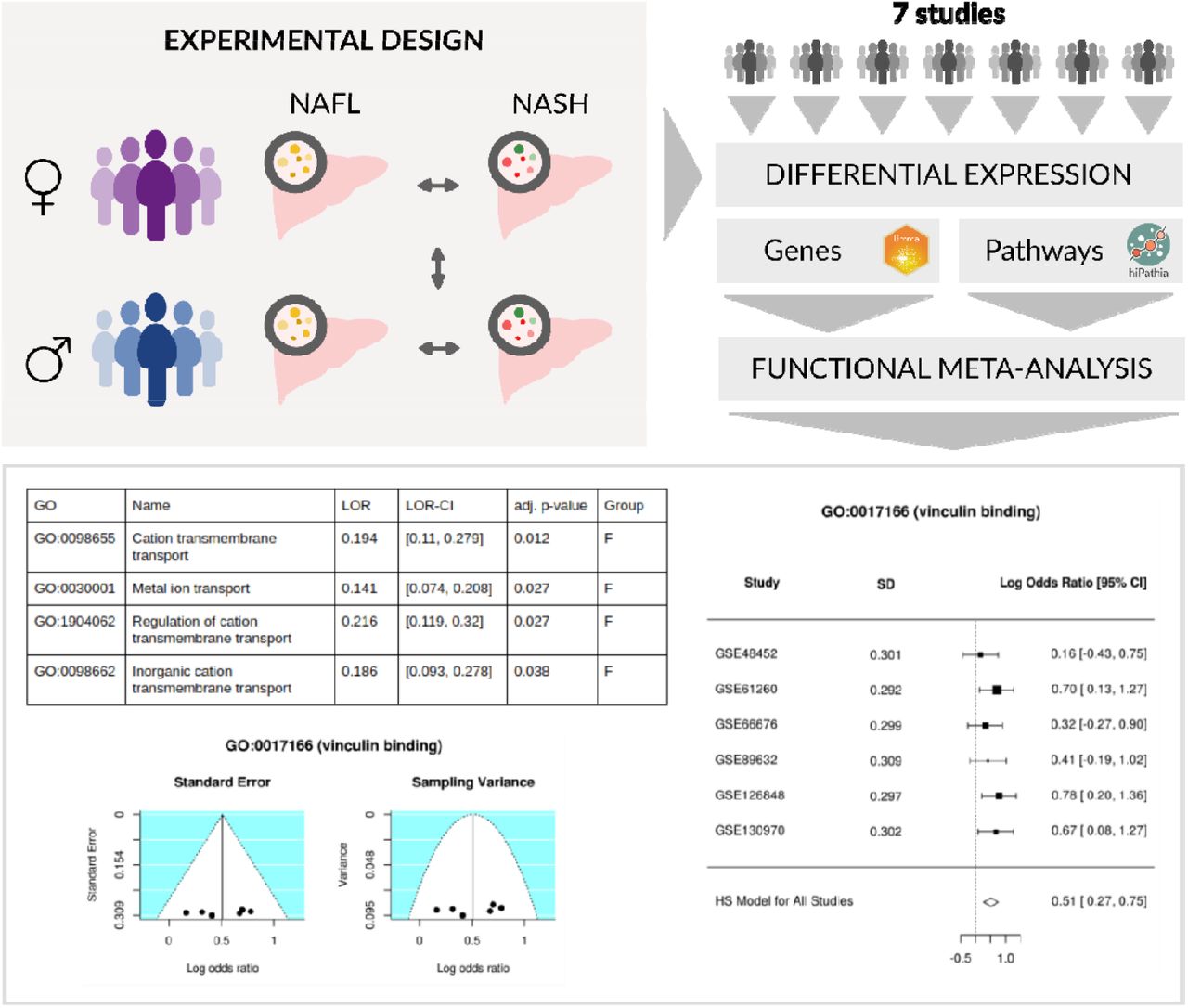
We applied the same three-step transcriptomic analysis strategy to each separate study: (i) data acquisition and preprocessing; (ii) differential gene expression and functional enrichment analysis; (iii) differential pathway activation and functional analysis. The functional results of all the studies were then integrated using meta-analysis techniques (Figure 2). This work was carried out using the programming language R 3.6.0 [13]; information about the packages we used and their version numbers are provided in the supplementary material.
Data acquisition and preprocessing
During the data preprocessing we standardised the nomenclature of the different stages of the disease (by grouping the patients into Control, NAFL, and NASH groups) as well as the association of the probe identifiers with their corresponding genes. For studies that had used microarrays, the probe codes were transformed to their respective Entrez identifiers from the NCBI database [14].
For repeated probes, we calculated the median of their expression values. When available, data normalised by the original authors of the studies were used. Otherwise (studies GSE61260 and GSE66676), the raw data was normalised using the Robust Multichip Average (RMA) algorithm [15]. The gene nomenclature in RNA-Seq studies was also standardised to the Entrez identifiers and the raw data matrix was processed using the TMM standardisation method [16] followed by a log-transformation of the data.
To meet the objective of this work, women were separated into premenopausal and postmenopausal groups. Since the selected studies did not include this information, we assumed that women aged under 50 years had been premenopausal, based on previous literature indicating that the average age of menopause is around 48–52 years [17,18].
After the normalisation of the data we attempted to detect any possible anomalous effects within the studies by completing an exploratory clustering and a principal component analysis (PCA).

Differential gene expression and functional enrichment analysis
We carried out a differential expression analysis of these studies using the limma and edgeR packages [19,20] to detect genes that were differentially expressed during the progression of NAFL to NASH in premenopausal women versus men. For each gene, we adjusted a linear model, which included possible batch effects, contrasting (NASH.W – NAFL.W) and (NASH.M – NAFL.M), where NASH.W, NAFL.W, NASH.M, and NAFL.M corresponded to NASH-affected premenopausal women, NAFL-affected premenopausal women, NASH-affected men, and NAFL-affected men, respectively. The statistics of the differential expression were calculated and the p-values were adjusted using the Benjamini & Hochberg (BH) method [21].
Next, based on the differential gene expression results, we performed a functional enrichment analysis using gene set analysis (GSA) [22]. First, the genes were ordered according to their p-value and the sign of the contrast statistic. Next, the GSA was performed using the logistic regression model implemented in the mdgsa R package [23] along with their corresponding functional annotations obtained from the Gene Ontology (GO) [24] and Kyoto Encyclopaedia of Genes and Genomes (KEGG) PATHWAY [25] databases.
Because of their hierarchical structure, the gene annotations with GO terms applied in the mdgsa package were propagated so that they inherited the annotations of the ancestor terms. Excessively specific or generic annotations (blocks smaller than 10 or larger than 500 words) were subsequently filtered out. Finally, functions with a BH-adjusted p-value under 0.05 were considered significant.
For the two functional elements (GO terms and KEGG paths), we analysed the number of over-represented elements shared by the studies. These results were graphically represented as UpSet plots [26] to show the number of elements in common between the different sets. First, we compared the overrepresented elements as separate graphs to detect the common functions in each group: GO terms in premenopausal women, GO terms in men, KEGG pathways in premenopausal women, and KEGG pathways in men. We then visualised any significant GO terms in each of the individual studies in the same plot to highlight significant terms with different signs from among the studies.
Differential pathway activation and functional analysis
The Hipathia algorithm [27] was used to perform Pathway Activity Analysis (PAA). This method transforms gene expression values for stimulus-response signalling sub-pathways into the activation levels which ultimately trigger cellular responses. In this current work we used the hipathia R package to analyse 1,654 GO terms and 142 UniProt functions [28], associated with 146 KEGG routes [25].
The activation signal of each subpathway was computed from gene expression values using this algorithm. These values were used to detect significant differential activations in the (NASH.W – NAFL.W) – (NASH.M – NAFL.M) contrast pair (as defined above). The functional annotations included in Hipathia and the differential activation results were integrated using the GSA method to identify differences at the functional level (GO terms and UniProt functions) in relation to progression of the disease.
Meta-analysis
Once the gene functional analysis was applied to each individual study, we carried out a functional meta-analysis to summarise the results. Similarly, once the pathway functional analysis was applied to each individual study, the results were summarised via a homologous functional meta-analysis.
Both meta-analyses were performed following the methodology described in detail in García-García [29]. Briefly, the metafor R package [30] was used to evaluate the combined effect with a random-effects model; this model more precisely detects overrepresented elements than performing the studies individually at a higher statistical power. Likewise, variability in the individual studies was incorporated into the global estimation of the measured effect so that less-variable results had a higher weight in the overall calculation of the logarithm of the odds ratio (LOR). Finally, we confirmed the suitability of the selected studies by evaluating the heterogeneity of these indicators.
For each meta-analysis and function, p-values (adjusted using the BH method), LORs, and 95% confidence intervals (CIs) were calculated. A term was considered significant when its adjusted p-value was lower than 0.05; a positive LOR in significant functions indicated a greater overrepresentation in premenopausal women than in men. In contrast, a negative LOR indicated higher representation in men than in premenopausal women. Finally, funnel plots and forest plots were used to assess the variability and to measure the contribution of each study to the meta-analysis.
A total of 8,223 elements were analysed in the gene functional meta-analysis (7,994 GO terms and 229 KEGG pathways); 1,654 GO terms and 142 UniProt functions were assessed in the functional pathway meta-analysis [28] in association with 146 KEGG pathways [25].
Results
Systematic review and exploratory analysis
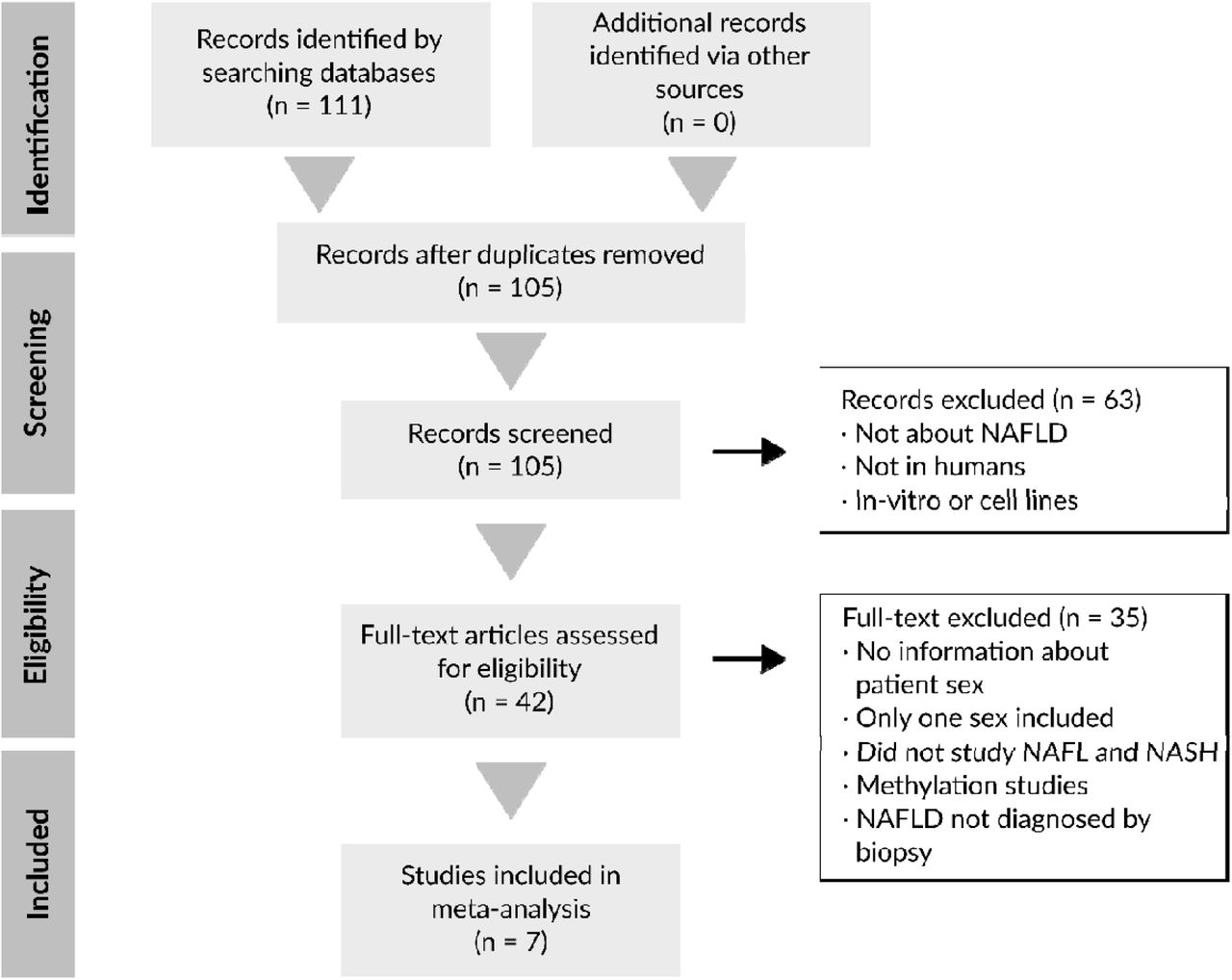
As shown in the PRISMA flow diagram (Figure 2), our systematic review yielded 105 non-duplicated studies. After applying the exclusion criteria described above, a total of 7 studies were included in this work. Studies were excluded for the following reasons: studies not in humans or unrelated to NAFLD (n = 48), in vitro studies (n = 15), lack of information on sex or inclusion of only one sex (n = 19), no patients had the NAFL and NASH disease stages (n = 11), and diagnoses not based on histology or methylation studies (n = 5).

The 7 selected studies included a total of 624 patients (Supplementary Table 1); 253 individuals were men and 371 women (40.5% men and 59.5% women), of which 253 were aged under 50 years and 118 were older (68.2% and 31.8%, respectively). By disease stage, 195 belonged to the control group, 166 to NAFL, 234 to NASH, and 29 could not be classified because this information was not available (31.25%; 26.60%, 37.50%, and 4.65%, respectively).
Of note, although the GSE83452 study contained patients at baseline and at one-year post-treatment, only baseline individuals were used in this current study. Table 1 shows the number of individuals from each study that were eligible in our work based on the criteria we established. Our exploratory analysis showed abnormal behaviour for the GSE83452 study; the PCA showed two different blocks: the first 64 samples were grouped into one cluster and the last 88 into another one. We considered this a batch effect and took it into account in our subsequent analyses.
Individual analysis
The functional analysis of individual genes highlighted significant results overrepresented in both the groups of premenopausal women and of men, as summarised in Table 2. Analysis (using UpSet plots) of the terms or pathways that were common through the different studies showed that none of the elements were common to more than 4 studies (Supplementary Figure 1); in fact, the results for some elements were opposite in different studies (Figure 3a). The functional analysis of individual pathways only exhibited significant results for the GSE83452 study (20 sub-paths and 96 GO terms).
Positive and negative logarithm of the odds ratio (LORs) represent overrepresentation in premenopausal women and in men, respectively.
Meta-analysis
In addition to the p-value associated with each of the GO terms, the results of the meta-analysis measured the overall effect of that term throughout all the individual studies, i.e., the LOR. This effect indicated the magnitude of the overexpression of each term (the LOR value) and its direction (the LOR sign). Figure 3b shows, for one function, the LOR of each individual study and the joint LOR of the meta-analysis. The results of all significant elements are detailed in the Supplementary Figures 2 and 3. We confirmed the absence of bias and heterogeneity in all cases, as in Figure 3c (Supplementary Figures 4 and 5).

(A) UpSet plot showing the number of common elements among the significant GO terms in our functional gene analysis. Only the 20 most abundant interactions are shown. Horizontal bars indicate the number of significant elements in each study; the two groups (premenopausal women and men) are separated by a red line. The vertical bars indicate the common elements in the sets, indicated with dots under each bar. The single points represent the number of unique elements in each group. Cases which cross the red line are significant GO terms, both in premenopausal women and men. (B) A forest plot of the GO:0017166 term, showing the LOR of each study and the global result. (C) Funnel plot of the GO:0017166 term; dots in the white area indicate the absence of bias and heterogeneity.
Gene functional analysis
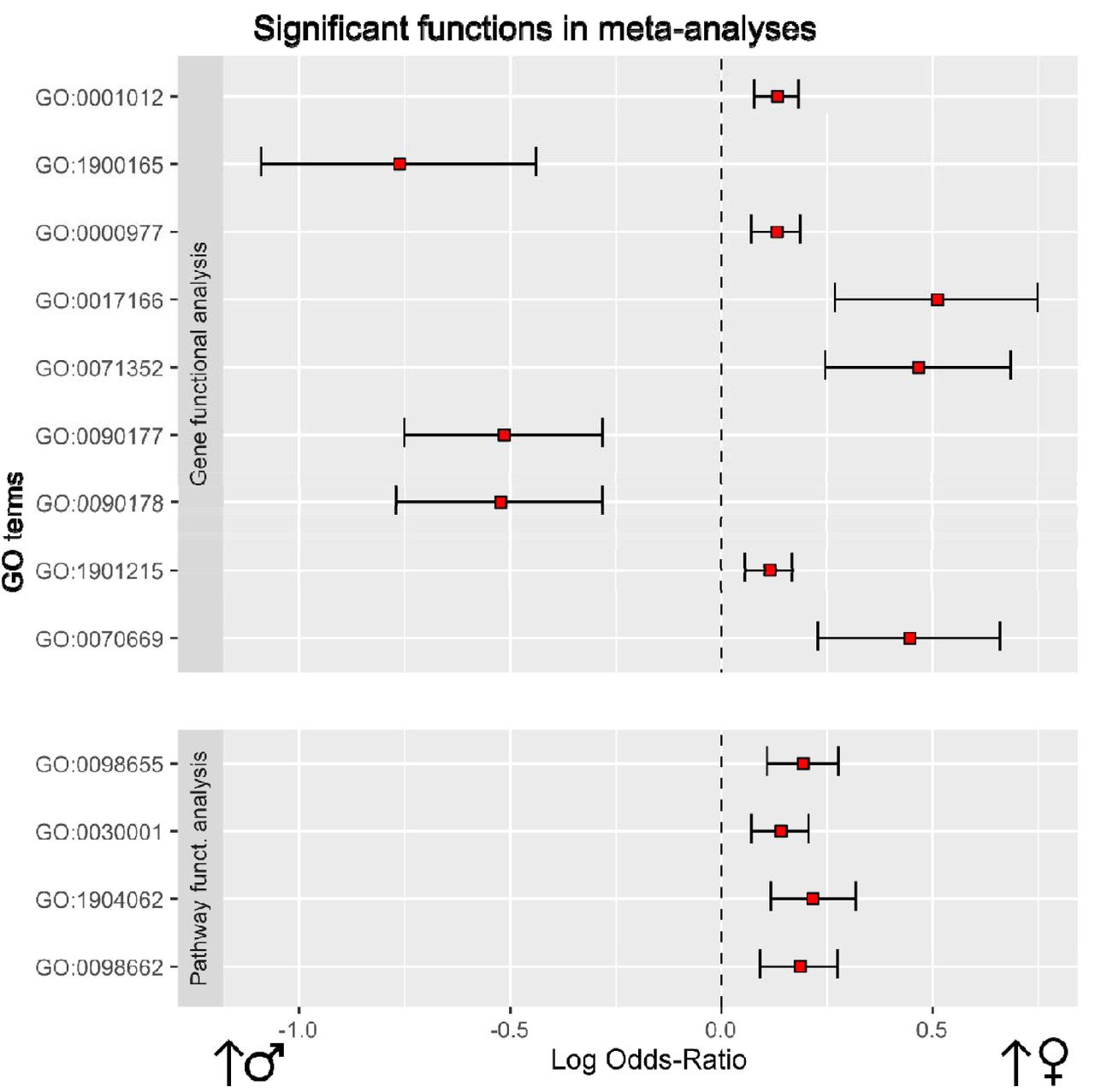
The functional meta-analyses of the individual gene differential-expression pipeline results showed a total of 9 significant GO terms (Table 3 and Figure 4), although no KEGG pathways were significant. Among the significant GO terms, 6 were overrepresented in premenopausal women; these elements encompassed a broad spectrum of biological processes and molecular functions, including RNA polymerase II regulatory region DNA binding (GO:0001012), RNA polymerase II regulatory region sequence-specific DNA binding (GO:0000977), Vinculin binding (GO:0017166), Cellular response to interleukin-2 (GO:0071352), Negative regulation of neuron death (GO:1901215), and Response to interleukin-2 (GO:0070669). The 3 remaining terms were overrepresented in men: Negative regulation of interleukin-6 secretion (GO:1900165), Establishment of planar polarity involved in neural tube closure (GO:0090177), and Regulation of establishment of planar polarity involved in neural tube closure (GO:0090178).
The logarithm of the odds ratio (LOR), its confidence interval (LOR-CI), the adjusted p-value, and the group in which the function was overexpressed are shown.
(W: women; M: men).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This plot shows significant functional terms of each meta-analysis. On the right: biological functions more overrepresented in women. On the left: biological functions more overrepresented in men. For each function, LOR (red square) and its confidence interval have been represented.
Pathway functional analysis
The functional meta-analysis of the individual results in the pathway differential-activation pipeline returned a total of 4 significant GO terms, all of them overrepresented in the progression of NAFLD in premenopausal women and with a LOR value lower than 0.25 (Table 3 and Figure 4). All these terms were related to the transport of ions and cations: Cation transmembrane transport (GO:0098655), Metal ion transport (GO:0030001), Regulation of cation transmembrane transport (GO: 1904062), and Inorganic cation transmembrane transport (GO:0098662). The meta-analysis of the UniProt functions did not provide any significant results.
Discussion
Although previous studies have analysed the molecular mechanisms involved in NAFLD, they had not usually considered gender and sex differences and so, sex-related molecular dissimilarities in this pathology remain unclear. Epidemiological studies have shown a higher prevalence of NAFLD in men and postmenopausal women compared to premenopausal women [3]. In order to unveil the molecular basis of these sex differences, here we present a systematic review and meta-analysis of publicly available genomic studies which had analysed differences in NAFLD disease progression between premenopausal women and men at a functional level.
The individual results of the selected studies revealed that no GO or KEGG terms were common across all 7 of the studies we selected. In fact, as shown in Figure 3, the results seem to contradict themselves because the same functions appear to be overexpressed in both groups, depending on the study. These results support the concept that meta-analyses, such as this one, are necessary to detect relevant and consistent knowledge when evaluating sets of studies. We identified a total of 13 affected GO terms through via two different functional meta-analysis approaches, based on differentially expressed genes or differentially activated pathways, respectively; these terms belonged to different types of functions related to inflammation, cell binding, and the establishment of polarity during neural tube closure.
Functional gene meta-analysis reported a total of 9 significant GO terms. Of these, the overrepresentation of Vinculin binding term (GO:0017166) has previously been reported in premenopausal women when compared to men. This cytoplasmic actin-binding protein is enriched in focal adhesions and adherens junctions, contributes to junction stability [38], and plays an important role as a regulator of apoptosis [39]. Of note, Arendt et al. previously found genes related to cell-cell adhesion (ANXA2 and MAG) that were differentially expressed between patients with NAFL and NASH, but their analysis did not separately consider men and women [40].
Our analysis highlighted differences between the sexes related to the expression of cytokine-related terms. Negative regulation of interleukin-6 secretion (GO:1900165) was overrepresented in men, while Cellular response to interleukin-2 (GO:0071352) and Response to interleukin-2 (GO:0070669) were overrepresented in premenopausal women. Interleukin-2 (IL-2) is well known to promote tolerance by regulating T-cell central tolerance, the formation of regulatory T cells, and activation-induced cell death [41,42]. Interestingly, IL-2 signalling is deficient in many chronic liver diseases [43]. IL-2 receptors are also expressed by B-cells and cells of the innate immune system [44]; yet, the role of IL-2 in human NAFLD remains poorly understood.
Interleukin-6 (IL-6) is also a well-recognised regulator of hepatic regeneration [45,46] which has a complex role in liver pathologies, although its participation in the development of NAFLD remains unclear [47]. IL-6 may enhance hepatic repair and regeneration, however, it might also be involved in inducing insulin resistance and stimulating hepatocyte apoptosis, thus contributing to the development of NAFLD [48,49]. Indeed, Wieckowska et al. found a positive correlation between IL-6 expression in hepatocytes, and the degree of inflammation and the stage of fibrosis in patients with NAFLD [50].
Changes in the immune responsiveness between men and women may be one possible explanation for the sex-differences observed in NAFLD. These data suggest that women may have an immune-tolerant response to lipotoxicity, perhaps as a result of enhanced IL-2 signalling. Whereas, men may have an impaired liver-healing response to chronic injury as a result of poor IL-6 signalling. Nonetheless, these ideas remain unsubstantiated and will require further examination and validation in larger studies.
A recent study by Herrera-Marcos et al. detected sex differences in the control of the Cidec/Psp27β gene at the level of the liver [51]. In fact, expression of the mRNA of this gene has been correlated with the presence and density of liver lipid droplets under certain circumstances. Among other functions, the GO notation for Cidec/Psp27β is related to Negative regulation of neuron death (GO:1901215). Therefore, the fact that the result for this function was significant in our study could be related to the differences detected by Herrera-Marcos et al (2020).
Interestingly, we found other significant GO terms that had not been directly associated with NAFLD in previous studies. For instance, Establishment of planar polarity involved in neural tube closure (GO:0090177) and Regulation of establishment of planar polarity involved in neural tube closure (GO:0090178) were overrepresented in men with a LOR exceeding 0.5 (−0,514 and −0,523). However, we did not find a direct relationship between these terms and NAFLD itself. Given that the core planar cell polarity (PCP) module comprises six proteins localised in adherens junctions [52], these terms could be related to cell-cell junctions or inflammation.
Curiously, we did not find references to RNA polymerase II regulatory region DNA binding (GO:0001012) and RNA polymerase II regulatory region sequence-specific DNA binding (GO:0000977) in the literature related to NAFLD. Our functional pathways meta-analysis showed 4 significant GO terms involved in cation transport: Cation transmembrane transport (GO:0098655), Metal ion transport (GO:0030001), Regulation of cation transmembrane transport (GO:1904062), and Inorganic cation transmembrane transport (GO:0098662). The alteration of pathways and functions related to cations have been described previously for NAFLD [53], but information about their specific involvement in this disease remains scarce.
Terms related to IL-2 and IL-6, such as Vinculin binding and Establishment of planar polarity involved in neural tube closure, showed LOR values close to or above 0.5, indicating a strong difference between the groups. In essence, our results show alterations in biological processes and molecular functions in the progression from NAFL to NASH which were significantly different between men and premenopausal women. These elements may be helpful to better understand sex-related differences in NAFLD and their in-depth study could open the door to more personalised treatments for this pathology in the future.
Conclusions
The different clinical course and pathogenic mechanisms involved in NAFLD in men versus women were previously known but have been little studied. In order to try to better understand these differences, we revised 105 GEO studies and selected 7 suitable for a functional meta-analysis. We identified a total of 13 significant GO terms from these data, some of which have been previously related to the disease, thus indicating that the progression of NAFLD may be better understood when the factor of sex is considered. However, other functional terms we identified have never been linked to this disease, suggesting that more analyses will still be required to corroborate and expand upon these findings. In conclusion, meta-analyses of transcriptomic data are a powerful approach to identify sex-related differences in the progression from NAFL to NASH.
Data Availability
This work uses data published in previous studies, which is available in the Gene Expression Omnibus repository.
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48452
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE61260
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66676
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE83452
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89632
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126848
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130970
Conflict of interest statement
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Financial support statement
This research was supported by the Principe Felipe Research Centre.
Author contributions
JFC analysed the data, MRH and FGG designed and supervised the analysis, JFC, MRH, and FGG wrote the manuscript, MRH designed the graphical abstract, MB and HM helped in the interpretation of the results, FGG conceived the work. All the authors revised the manuscript and added their suggestions.
Author names in bold designate shared co-first authorship
Acknowledgements
The authors would like to thank the Principe Felipe Research Centre (CIPF) for providing access to its computational cluster in order to carry out this work. This study was co-funded by European Regional Development Funds (FEDER) awarded within the Valencian Community 2014–2020. We would also like to thank Dr. Kristoffer T.G. Rigbolt for providing information about ages of the sample used in study GSE126848.
References
- [1].↵
- [2].↵
- [3].↵
- [4].↵
- [5].↵
- [6].↵
- [7].↵
- [8].↵
- [9].↵
- [10].↵
- [11].↵
- [12].↵
- [13].↵
- [14].↵
- [15].↵
- [16].↵
- [17].↵
- [18].↵
- [19].↵
- [20].↵
- [21].↵
- [22].↵
- [23].↵
- [24].↵
- [25].↵
- [26].↵
- [27].↵
- [28].↵
- [29].↵
- [30].↵
- [31].
- [32].
- [33].
- [34].
- [35].
- [36].
- [37].
- [38].↵
- [39].↵
- [40].↵
- [41].↵
- [42].↵
- [43].↵
- [44].↵
- [45].↵
- [46].↵
- [47].↵
- [48].↵
- [49].↵
- [50].↵
- [51].↵
- [52].↵
- [53].↵



