
ABSTRACT
In May 2020 the Russian Ministry of Health granted fast-track marketing authorization to RNA polymerase inhibitor AVIFAVIR (favipiravir) for the treatment of COVID-19 patients. In the pilot stage of Phase II/III clinical trial, AVIFAVIR enabled SARS-CoV-2 viral clearance in 62.5% of patients within 4 days, and was safe and well-tolerated.
INTRODUCTION
The pandemic of the novel coronavirus infection (COVID-19) represents an unprecedented disaster for healthcare providers and economy worldwide. The urgent requirement for the effective treatments has sparked an intense effort on the part of the pharmaceutical industry and shifted drug development to a new scale of commitment and collaboration.
On May 29, 2020 the Russian Ministry of Health (MoH) granted fast-track marketing authorization to AVIFAVIR (favipiravir) for the treatment of COVID-19 patients. The drug was developed and brought to market within a period of only two months, a feat made possible by the highly concerted and coordinated efforts of regulator, industry, and health professionals. The marketing authorization makes AVIFAVIR the only oral drug approved for treatment of moderate to severe COVID-19 to date.
Favipiravir is an RNA-dependent RNA polymerase inhibitor marketed in Japan (Avigan) [1] and China (Favilavir) as a second-line treatment of novel or re-emerging influenza outbreaks. Earlier this year it was reported to demonstrate antiviral activity against SARS-CoV-2 in Vero E6 cells (EC50, 61.88 μM, CC50 > 400 μM, SI > 6.46) and to provide shorter viral clearance time in patients with COVID-19 [2]. A non-randomized study conducted in China demonstrated a median viral clearance time of 4 days, versus a period of 11 days for lopinavir/ritonavir (p < 0.001) [3]. The World Health Organization (WHO) also listed Favipiravir as a candidate experimental treatment (broad spectrum antiviral) [4].
In the attempt of establishing COVID-19 treatment in the Russian Federation, in March 2020, the Russian Direct Investment Fund and ChemRar Group organized a joint venture aimed at research, development, manufacturing, and marketing of favipiravir under the brand name AVIFAVIR. The API synthesis and the final drug product manufacturing were performed at the Good Manufacturing Practice facility of Chemical Diversity Research Institute.
METHODS
Study design and participants
The pilot stage of the adaptive, multicenter, open label, randomized, Phase II/III clinical trial of AVIFAVIR versus standard of care (SOC) in hospitalized patients with moderate to severe COVID-19 pneumonia was conducted by IPHARMA CRO in April and May 2020 at six Russian clinical sites based at infectious hospitals in Moscow, Smolensk, and Nizhniy Novgorod (ClinicalTrials.gov Identifier: NCT04434248). The Independent Data Monitoring Committee (IDMC) was introduced to review the results of the interim analysis.
The objective of the pilot stage of the clinical study was to conduct a preliminary assessment of the efficacy and safety of AVIFAVIR, and to select the optimal dosing regimen for further evaluation in moderately ill SARS-CoV-2 virus-infected patients at the pivotal stage. The study design was based on WHO R&D Blueprint recommendations for clinical trials of therapeutics for COVID-19 [5].
Upon signing the informed consent form and screening, 60 eligible patients with pneumonia associated with PCR confirmed COVID-19 were randomized at a 1:1:1 ratio to receive either AVIFAVIR 1600 mg BID on Day 1 followed by 600 mg BID on Days 2-14 (1600/600 mg), or AVIFAVIR 1800 mg BID on Day 1 followed by 800 mg BID on Days 2-14 (1800/800 mg), or SOC. Each treatment group comprised 20 patients and all randomized patients constituted safety and intent-to-treat (ITT) analysis sets.
In the 40 AVIFAVIR-treated patients, the drug was administered for a mean period of 11 days, with antibiotics (30%) and anticoagulants (35%) as concomitant medications for COVID-19. SOC was assigned in the control group according to the recommended treatment schemes presented in the Russian MoH interim guidelines for treatment of COVID-19 [6]. Patients received hydroxychloroquine (65%), chloroquine (10%), lopinavir/ritonavir (5%), or no etiological treatment (20%), with antibiotics (40%) and anticoagulants (20%) as concomitant medications for COVID-19.
The AVIFAVIR and control groups were generally comparable in demographic and baseline characteristics. 36.7% of patients (22/60) had risk factors for severe disease (age 60 and older and/or concurrent chronic conditions). At baseline, 73.3% of patients (44/60) were on ambient air (Score 3 on the WHO Ordinal Scale for Clinical Improvement) and 26.7% of patients (16/60) required supplemental oxygen via mask or nasal cannula (Score 4). Mean disease duration was 7 days from the start of the first symptoms. Most common symptoms were body temperature above 37.5°C (95.0%), cough (83.3%), weakness (70.0%), anosmia (35.0%), chest tightness (30.0%), and dyspnea (28.3%).
The study assessments included daily vital signs, SpO2 and WHO Ordinal Scale for Clinical Improvement; PCR for SARS-CoV-2 virus detection in nasopharyngeal/oropharyngeal swabs at baseline and on Days 5, 10, and 15; chest computed tomography (CT) scan at baseline and on Day 15; physical examination, complete blood count with differential, biochemistry, C-reactive protein, urinalysis, and electrocardiogram at baseline and on Days 5 and 15. If not discharged from the hospital earlier, the patients attended follow-up visits on Day 22 and Day 29. All patients were followed until Day 29.
RESULTS
Interim efficacy and safety results
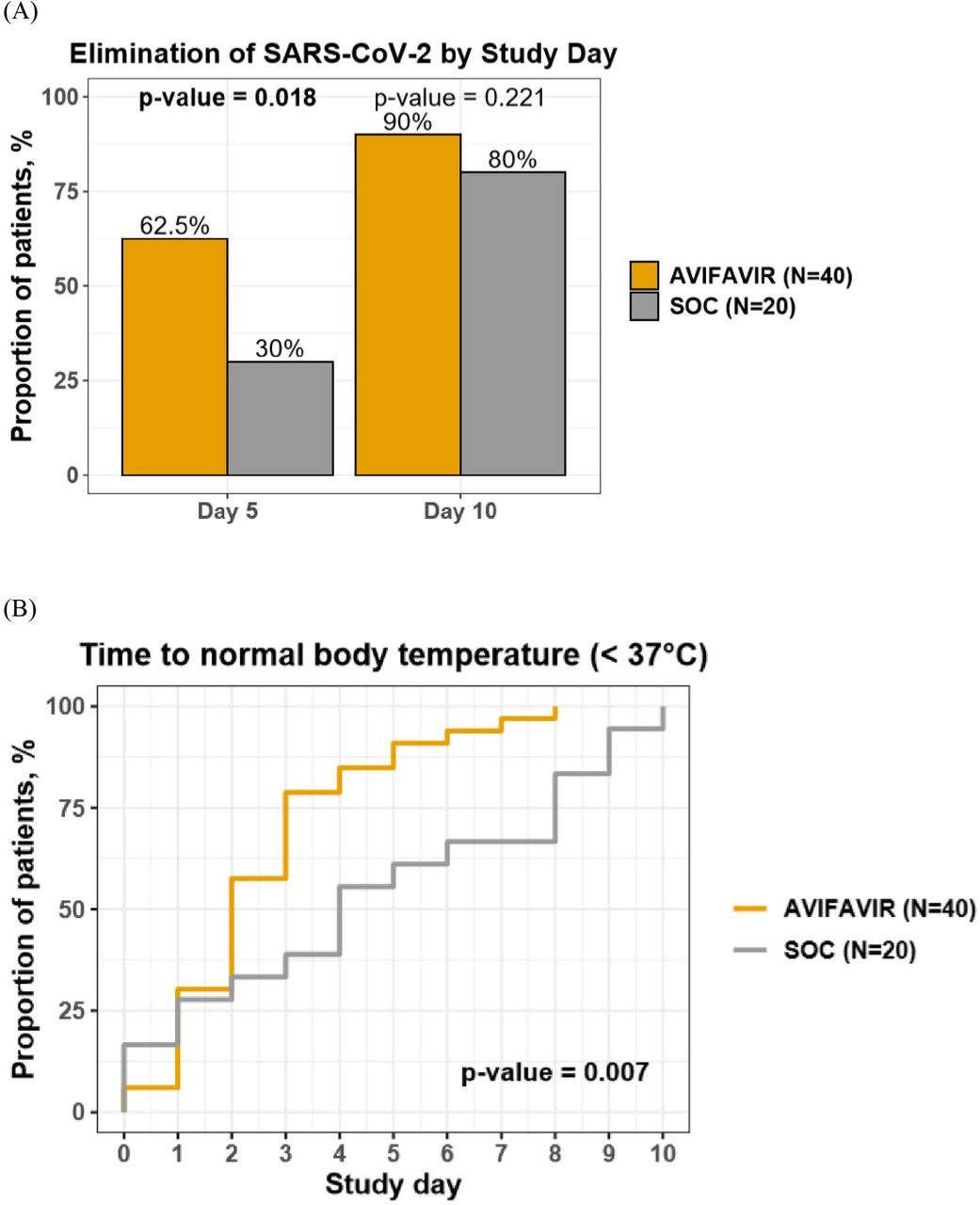
The primary efficacy endpoint at the pilot stage was the elimination of SARS-CoV-2 by Day 10 (defined as two negative PCR tests with at least a 24-hour interval). The “go-no-go” decision to start Phase III was based on the exact single-stage Phase II assessment at one-sided α=0.05 and 80% power [7]. Thirteen or more responders out of 18 treated patients would have been sufficient to demonstrate that the effect of the viral clearance in 80% of patients by Day 10 is plausible, and that the efficacy is greater than the presumably non-effective level of 50%.
On Day 5 (i.e. 4 days after treatment was started), the viral clearance was achieved in 62.5% of patients (25/40) on AVIFAVIR (similar proportions on both dosage levels) and in 30.0% of patients (6/20) on SOC (p=0.018). By Day 10 (i.e. 9 days after treatment was started) the viral clearance was achieved in 90.0% of patients (36/40) on AVIFAVIR (similar proportions on both dosage levels) and in 80.0% of patients (16/20) on SOC (p=0.221).
The median time to body temperature normalization (< 37°C) was 2 days (IQR 1-3) in the AVIFAVIR groups and 4 days (IQR 1-8) in the SOC group (p=0.007). By Day 15, chest CT scans improved in 82.5% of patients (33/40) on AVIFAVIR and 75.0% of patients (15/20) on SOC (p=0.494).
Adverse drug reactions to AVIFAVIR were reported in 17.5% of patients (7/40) including diarrhea, nausea, vomiting, chest pain and an increase in liver transaminase levels (ALT and AST). The adverse drug reactions were mild to moderate and caused early discontinuation of the study drug in 5.0% of patients (2/40).
During the pilot stage of the study, 2 patients on AVIFAVIR 1600/600 mg were moved to ICU and received mechanical ventilation, 1 of them died due to deterioration of the disease. Both patients had concurrent chronic conditions that increased the risk of severe disease.
By the time of the interim analysis 80.0% of patients (32/40) from the AVIFAVIR group and 90.0% of patients (18/20) from the SOC group were discharged from hospital after improvement in their condition.
Dose-response analysis
Two AVIFAVIR regiments (1600/600 mg and 1800/800 mg) demonstrated similar results in most efficacy and safety endpoint assessments. The median loading dose of AVIFAVIR was 43.9 mg/kg (IQR 40.0-47.1) in patients with confirmed negative PCR on Day 5, and 39.1 mg/kg (IQR 35.6-43.9) in patients with positive PCR on Day 5. The confirmed SARS-CoV-2 virus elimination on Day 5 on AVIFAVIR < 43 mg/kg was achieved in 47.3% of patients (9/19) and on AVIFAVIR ≥ 43 mg/kg was achieved in 80.0% of patients (16/20) (p=0.0362). No dose-dependent toxicity was observed in the study.
Thus, 3 weight-based AVIFAVIR dosing regimens were suggested for the pivotal stage of the study. The recommended duration of AVIFAVIR treatment was 10 days, although it can be reduced based on laboratory-confirmed viral clearance.
CONCLUSIONS
At the pilot stage of the Phase II/III open label, randomized clinical trial in the Russian Federation, AVIFAVIR therapy demonstrated rapid antiviral response against SARS-CoV-2. 62.5% of AVIFAVIR-treated patients (25/40) achieved viral clearance within 4 days after initiation of therapy, which was significantly higher than in the SOC group (p=0.018). There were no new safety concerns related to AVIFAVIR as all adverse reactions were mild to moderate in severity and were consistent with those reported previously for AVIGAN [1, 8]. Dose-dependent efficacy pattern was identified and will be studied further. The results of the pivotal stage of the study are expected in July 2020.
The fast-track marketing authorization granted by the Russian MoH makes AVIFAVIR the only approved oral drug for treatment of moderate to severe COVID-19 to date.
Data Availability
N/A
Financial support
This project is supported financially by the Russian Direct Investment Fund, the Ministry of Industry and Trade of the Russian Federation and The Skolkovo Innovation Center.
Potential conflicts of interest
None declared.
Authors’ contributions
EPP, NVL, EAS, IGG, ENS, EGT were Principal Investigators responsible for patients’ recruitment, study treatment, and data collection in compliance with the Protocol and ICH GCP. AAI, KAD and TAS conceived this project, proposed a variant of its organization and controlled the progress of its implementation. NVV, VNA and ANE developed a clinical trial protocol, worked on the statistical aspects of the study and the analysis of the results. EAM, AAB and EVY organized clinical trials and collection of the results. RNK developed a preclinical study design and organized its implementation. API, DVK developed the technology and organized the production of the substance. NAP has developed the composition and technology for the production of AVIFAVIR tablets. NPS studied and analyzed a possible market for AVIFAVIR. AVI carried out scientific management of the project and edited Rapid communication.

{kind=link}
A. Elimination of SARS-CoV-2 virus in nasopharyngeal/oropharyngeal swabs of patients treated with AVIF AVIR or SOC on Day 5 and Day 10 of study treatment. B. Time to normal body temperature in patients treated with AVIFAVIR or SOC (Kaplan-Meier curves).
Acknowledgements
The authors thank the co-investigators MD Oksana Y. Shaydyuk of City Clinical Hospital n.a. O.M. Filatov (Moscow, Russia); MD, PhD Dmitry A. Garkavi and MD Anna V. Bushmanova of First Moscow State Medical University n.a. I.M. Sechenov (Moscow, Russia); MD, PhD Anton V. Potapenko and MD Anna G. Sorokina, Moscow State University n.a. M. V. Lomonosov (Moscow, Russia); MD PhD Olga S. Rozinkova of Clinical hospital No.1 (Smolensk, Russia); MD, PhD Veronika S. Vasilieva, MD Ekaterina E. Shokhina, MD Natalia O. Simonova, and MD Valery A. Luybinskiy of Central Clinical Hospital with Polyclinic (Moscow, Russia) as well as Independent Data Monitoring Committee, including MD, PhD, Professor Alexey V. Kravchenko of Central Research Institute of Epidemiology (Moscow, Russia), MD, PhD, Professor, Russian Academy of Sciences Professor Kirill A. Zykov of Moscow State Medical Dental University n.a. A.I. Yevdokimov (Moscow, Russia) and MD, PhD, Professor Vladimir V. Rafalskiy of Immanuel Kant Baltic Federal University (Kaliningrad, Russia).



