
Abstract
Male Breast Cancer (BC) is relatively rarer, accounting for less than 1% of cancers in men. MBC is hereditary in nature and mainly attributed to BRCA1/2 germline mutations. Accordingly, National Comprehensive Cancer Network (NCCN) guidelines advise genetic counselling and testing for all cases of MBCs and their unaffected family members. In this report, we present an uncommon case of male patient primarily diagnosed with pancreatic cancer who later developed asynchronous bilateral hormone positive breast cancer. We describe the genetic screening and clinical management protocol for the proband and family members. Genetic testing with next generation sequencing by uses of a multi-gene germline mutation panel revealed a likely pathogenic BRCA2 variant (c.8754G>A, p.E2918E). Subsequently, 34 members of the extended family of the proband were tested for the BRCA2 variant by Sanger sequencing. 6 of the family members were identified as carriers of this BRCA2 variant. Of these, three presented with hereditary breast cancer and 3 were unaffected healthy carriers. In silico analysis for mechanistic insights in underlying pathogenicity revealed that the silent BRCA2 mutation is a spliceogenic variant that is likely to create an aberrant mRNA transcript via alternative splicing of BRCA2 gene. Our study demonstrates the clinical relevance of this silent BRCA2 mutation and emphasizes the need for further experimental studies to elucidate its functional role in breast cancer pathology.
Introduction
Male Breast Cancer (MBC) is a rare disease accounting for 1% of the total breast cancers in the world (Moelans et al., 2019). The lifetime risk of a man developing breast cancer is 1:1000 (100 times less than that in a woman) (Abdelwahab Yousef, 2017). The risk of developing male breast cancer increases linearly after the age of 60 years and peaks around 70 years (Vietri et al., 2020) and is generally observed in older men. Given the rarity of the disease and lack of large cohorts, the recommendations and clinical management of the disease today, are mainly based on the data extrapolated from female breast cancer studies.
Demographic, environmental, epidemiologic and genetic factors are risk factors known to influence the onset of MBC (Weiss et al., 2005). Elevated levels of estrogen prior to radiation exposure and strong family history also contribute to the increased MBC risk (Piscuoglio et al., 2016). The NCCN guidelines for genetic testing of hereditary breast, ovarian, prostate and pancreatic cancers have suggested genetic testing in a breast cancer affected male as well as his 1st degree relatives. MBCs are most frequently found to be Infiltrating Ductal Carcinoma and generally present as a lump in the breast. The staging of the cancer reflects the degree of tumour spread and the prognosis, in a fashion similar to breast cancer in females (Fentiman, 2016).
Pathogenic germline mutations in the high penetrance BRCA1 and BRCA2 genes, which act as tumour suppressors, have been long-established as the key mediators of predisposition to hereditary breast and ovarian cancers (Pfeffer et al., 2017). Functionally, BRCA1 and BRCA2 are key players in double stranded DNA repair pathways and maintain genomic integrity through homologous recombination during replication. Germline BRCA1/2 mutations account for 25% of hereditary breast and ovarian cancer (HBOC). BRCA2 contributes to about 6-10% of mutations in male breast cancer. Recent reports indicate that BRCA1/2 mutations also increase the risk of developing pancreatic and prostate cancers (Ibrahim et al., 2018; Kast et al., 2016; Nielsen et al., 2016; Shah et al., 2020; Tripoli & Cordova, 2020).
The BRCA1 gene with 24 exons, encodes an 1863 amino acid protein and is most frequently mutated in N-terminus RING domain, exon 11-13 and C-terminus BRCT domain (Pfeffer et al., 2017). The BRCA2 with 27 exons, encodes for a 3418 amino acid protein and is characterised by the BRC repeat, a conservative segment, coded by exon 11, and a C-terminus oligonucleotide binding (OB) region (Venkitaraman, 2019).
Unlike the breast cancers in females, the patho-etiology of MBCs has not been well studied. Recent reports have highlighted an important role for BRCA1/2 germline mutations in predisposition towards MBC. Population genetic studies have revealed that approximately 0-4% men diagnosed with breast cancer harbour a BRCA1 mutation and 4-16% of them harbour a BRCA2 mutation (Ferzoco & Ruddy, 2015). When compared with female breast cancers, BRCA2 mutations are more common in MBCs than BRCA1 mutations indicating the potential difference in etiology of breast cancers in males and females (Fentiman, 2016). MBCs with a BRCA2 mutation present a significantly higher grade of breast cancers than BRCA2 mutated cancers in females. The presentation of MBCs at an advanced stage is presumed to be due to lack of mammographic screening programs for men and the lack of awareness about MBCs (Silvestri et al., 2016).
In this report, we present a case of a male patient (proband) initially diagnosed with pancreatic cancer and subsequently diagnosed with asynchronous bilateral breast cancer. Genetic testing during clinical management identified the proband as a carrier of a BRCA2 likely pathogenic variant (c.8754, G>A, p.E2918E). We present the clinical management and genetic screening approach for familial genetic risk assessment in the proband and his family. Furthermore, we provide insights into the potential biological mechanisms underlying the pathogenicity of this uncommon variant causing a synonymous change in BRCA2 protein (p.E2918E).
METHODOLOGY
Data Collection
Detailed compilation of clinical meta-data of the male patient (proband) was undertaken from our clinic. As per NCCN guidelines, 34 consenting members of the extended family of proband, 2 of which were breast cancer affected, were included in familial genetic risk assessment protocol. Family pedigree and medical history was obtained from the family members. Raw data for sequencing was obtained from partner genetic testing laboratory while the clinicopathology features were obtained from the medical records. Written informed consent was obtained from all the individuals. The study was approved by the Institutional Ethics Committee of PCCM (DCGI/CDSCO Registration Number: ECR/298/Indt/MH/2018. Every participant gave a separate informed consent for the use of their personal and medical information associated with this study. The study follows principles of the declaration of Helsinki (Brazil, 2013 version) and National Ethics Guidelines 2017 issued by the Indian Council of Medical Research, Government of India.
Clinical Management
The proband reported to our clinic with an initial diagnosis of pancreatic cancer and underwent clinical management in partner hospital. Breast Cancer diagnosis was based upon clinical examination and radiological evaluation of breast and axilla using CT abdomen 3 phase/ pelvis, Full Field Digital Mammography (FFDM) with 3-D Tomosynthesis (Siemens Mammomat Inspiration), Ultrasound, F18-FDG Whole Body PET CT scan and Ultrasonography (Siemens Acuson S2000). Histopathological studies on Tru-Cut biopsy on the sample was performed for confirming diagnosis of breast carcinoma. Similarly, ultrasonography and fine needle aspiration cytology were used for investigating axillary lymph node metastasis. The oncologic management with chemoradiation protocols was undertaken by a multidisciplinary clinical team in accordance with the current NCCN guidelines.
Genetic Risk Assessment
The pre and post-test counselling was performed as per the NCCN guidelines. After informed consent, the genetic testing was performed as per American College of Medical Pathologists (ACMG) guidelines in CAP-certified partner genetic testing laboratories. For familial risk assessment (i.e., cascade testing), individual counselling sessions were conducted prior to and after the cascade testing reports were available.
Genetic Testing
A. Next Generation Sequencing (NGS)
Genomic deoxyribonucleic acid (gDNA) was extracted from blood/saliva sample using standardized methodology (PrepIT-L2P kit from DNA Genotek, Canada) or QIAamp DNA Mini Kit (Qiagen, Germany) or the Nucleospin kit (Macherey–Nagel, Germany) and quantified by Qubit fluorimeter (Life Technologies). The next generation sequencing performed for the index patient included the genes associated with hereditary cancers and as per the ACMG guidelines. The TruSight Cancer Sequencing panel evaluated 30 genes associated with hereditary breast and endocrine cancers for the index patient. Next generation sequencing libraries were prepared by tagmentation, adaptor ligation, hybridization and were quantified to 6-10pM. Sequencing was performed using a standard v2 kit on Illumina MiSeq and the genetic variations were identified by using customised bioinformatics pipelines at partner genetic testing laboratory.
B. Sanger Sequencing
Sanger sequencing was performed in all 34 members of probands family for the variant c.8754G>A (p.E2918E), in exon 21 of the BRCA2 gene. Genomic DNA extracted from blood/saliva was processed using big dye terminator sequencing kit (Lifetech Inc., USA). Capillary electrophoresis performed on the Genetic Analyzer (3500 DX, Lifetech Inc., USA) was visualized using the Finch TV software (Geospiza, USA). Presence of the variant, c.8754G>A (p.E2918E), was evaluated by comparing the sample sequence with the reference sequence (RefSeq id: NM_000059).
In Silico Analysis
In silico splice site prediction tools namely NNSPLICE and Splice Port were utilized for bioinformatic analysis of spliceogenicity of the reported BRCA2 variant and associated pathogenicity.
For identifying population prevalence and clinical significance of the BRCA2 variant, database searches were undertaken for the reported BRCA2 variant. These included online databases namely, CIMBA (Consortium of Investigators of Modifiers of BRCA1/2); ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles); BCAC (The Breast Cancer Association Consortium); BIC (Breast Information Core); ClinVar and BRCA Exchange.
The family pedigree was constructed using Progeny (Progeny software LLC, South Bend, IN, USA).
RESULTS
Case Description
A male in his fifties presented with CT obstructive jaundice with peri-ampullary mass/ nodule measuring 1.4 x 2.2 cm. The mass was later confirmed to be pancreatic cancer by duodenoscopy-guided biopsy as a well differentiated adenocarcinoma of peri-ampullary region of pancreas. Whipple pancreaticoduodenectomy surgery was performed as part of clinical management.
A year later, the patient presented with a lump in one of his breast which was concluded to be benign epithelial lesions after a USG-guided FNAC procedure. However, the PET scan and subsequent biopsy revealed a Grade II Infiltrating Ductal Carcinoma. Immunohistochemical analysis identified the mass with an estrogen receptor (ER) / progesterone receptor (PR) positive (ER-80%, PR-70%), Human epidermal growth factor receptor (Her2-) negative status. Modified radical mastectomy was performed post primary diagnosis. The axillary excision biopsy revealed 2 out of 15 pathologically positive axillary lymph nodes, suggesting tumour metastases. Histopathology reports indicated moderately differentiated adenocarcinoma with perineural invasion and positive for Cytokeratin-7. Patient was treated with 6 cycles of 5-FluoroUracil; Epirubicin; Cyclophosphamide (FEC) adjuvant chemotherapy post-surgery. Subsequently, Linear accelerator based IMRT/3DCRT radiotherapy was administered. Hormonal therapy (Tamoxifen) was prescribed for 5 years after the completion of the treatment.
Two years later, the patient, presented with a bloody discharge from the nipple of the contralateral breast. On targeted sonomammography, two dilated ducts were seen at the 6 ‘o’clock position. The larger lesion was 2.7 mm in size. Internal echoes, increased peri ductal and internal vascularity was noted. Cytology of the nipple discharge of the was performed and was found to be positive for malignant cells. The biopsy findings and histological examinations characterised the breast tumour as Invasive ductal carcinoma Grade I, ER/PR positive. PET scan revealed a distant metastatic lesion in the left anterior abdominal wall measuring 61.5 x 29.9 mm. The patient was treated with Neoadjuvant hormone therapy (Anastrazole) and GnRH analogue which was followed by modified radical mastectomy.
In the same year, one of the relatives of the proband was diagnosed with ER/PR positive breast cancer (left). A few months later another relative was diagnosed with ER/PR positive breast cancer (right). In parallel, the proband was diagnosed with lung metastases and was treated with chemotherapy.
Considering the case history (i.e., pancreatic cancer followed by asynchronous bilateral breast cancer) was indicative of hereditary breast and ovarian cancer syndrome, the proband and family members were advised genetic testing. After written informed consent, the proband underwent a comprehensive multi-gene panel covering 30 genes associated with hereditary breast and ovarian cancer, including the 25 genes recommended by ACMG.
Genetic Analysis by Multi-gene Panel
Multi-gene panel testing by next generation sequencing included 30 high, moderate and low penetrance genes recommended by ACMG, predisposing to HBOC, pancreatic cancer and prostate cancer. Interestingly, only one heterozygous ‘likely pathogenic’ variant (c.8754G>A) on exon 21 of a high penetrance BRCA2 gene was reported out of the 30 genes tested.
The reported variant showed a heterozygous nucleotide change ‘G>A’ at position c.8754 in the BRCA2 gene and was also confirmed by sequencing with forward primer (flanking the BRCA2 variant) in two independent experiments. The detected variant resulted in no amino acid change (p.E2918E) when compared with the reference sequence (RefSeq ID: NM_000059). Since this variant was identified to cause a synonymous substitution in the BRCA2 protein (p.E2918E), it was referred to as a silent mutation.
Cascade Testing
As per NCCN guidelines, cascade testing of other affected and unaffected family members of the proband was performed to identify the risk of HBOC, pancreatic cancer and prostate cancer. After pre-test counselling, other family members (affected and unaffected) were tested for the previously identified BRCA2 mutation by Sanger sequencing (i.e, capillary electrophoresis on Genetic Analyzer).
Familial Risk Assessment
Considering the strong family history of pancreatic and breast cancer, the extended family of the proband underwent genetic testing after counselling and providing written informed consent. Among the 34 family members of the proband, six members were carriers for the identified BRCA2 variant (c.8754G>A). These include the relatives who were previously diagnosed with hormone positive breast cancer and 3 other unaffected family members.
Post-test Genetic Counselling
As per NCCN guidelines, the healthy unaffected BRCA2 carriers in the extended family of the proband were counselled by genetic counsellors to explain the risk of developing HBOC. Medical advice was provided by onco-clinicians about surveillance schedules, medication, and preventive surgeries as applicable.
Bioinformatic Analysis of BRCA2 variant (c.8754G>A)
The BRCA2 variant identified as c.8754G>A is located on chromosome 13q12.3. The dbSNP database has reported the identified variant as Synonymous Variant within the BRCA2 gene (rs80359803). The ClinVar database has reported this variant (RCV000077451.3) with a clinical significance and classified as ‘pathogenic’.
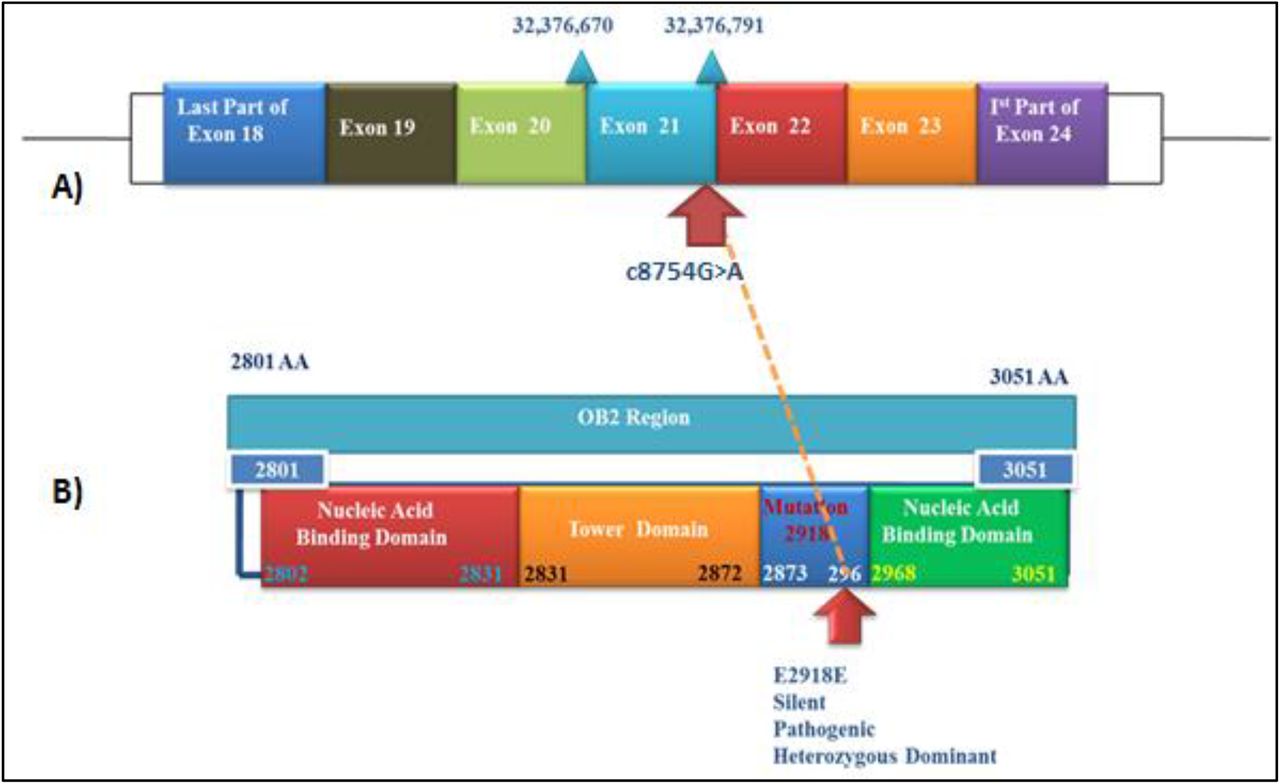
This variant (c.8754G>A) resides 1 base upstream of the splice donor site within exon 21 of the BRCA2 gene. Splicing prediction software programs such as NNSPLICE and Splice Port reveal that this variant is likely to affect splicing at the junction of exon 21 and intron 21 of the BRCA2 gene (i.e., spliceogenic) (Figure 2). This spliceogenic variant was located at conserved splice donor and acceptor sites. Evolutionary conservation analysis indicates that the Exon21-Intron junction is highly conserved among multiple species. According to the ACMG pathogenic variant classification standards, spliceogenic variants are known to induce major aberrations in the transcript, generate a premature termination codon or cause in-frame deletion of a known functional domain. Therefore, the identified spliceogenic BRCA2 variant was classified as likely pathogenic.

A) Sanger sequencing data of the proband harbouring the detected variant (c.8754G>A). A heterozygous nucleotide change ‘G>A’ at position c.8754 in the BRCA2 gene, clearly outlined on the electropherogram.
B) Sanger sequencing data revealing the individual was ‘positive’ (carrier) for the tested variant (c.8754G>A). The electropherogram clearly displays a heterozygous nucleotide change ‘G>A’ at position c.8754 in the BRCA2 gene.
C) Sanger sequencing data revealing the individual was ‘negative’ for the tested variant (c.8754G>A). The electropherogram clearly displays no change, with reference nucleotide ‘G’ being preserved at position c.8754 in the BRCA2 gene similar to the RefSeq.

{kind=link}
{kind=link}
A) The variant (c.8754G>A) is located 1 base upstream of the splice donor site within exon 21 of the BRCA2 gene.
B) Diagram representing the structure and domains of the OB2 region, contained within the DNA binding domain of the BRCA2 gene. This region harbours the identified silent mutation (p.E2918E). Arrows indicate the position of the identified variant.
BRCA2, protein is a 3,418 amino acid protein constituting an N-terminal transactivation domain (23–105), 8 central BRC repeats of ∼30 amino acids each (987–2113), and a Conserved C-terminal domain BRCA2, CTD (2479–3152). BRCA2 CTD has an α-helical domain and three Oligonucleotide/Oligosaccharide binding (OB) domains (OB1, OB2 and OB3). The OB2 domain (2801-3051) has multiple potential binding sites including ssDNA binding site, binding surface and interface.
At the protein level, this identified silent mutation (p.E2918E) resides in the OB2 (oligosaccharide binding fold module) region, which is contained within the DBD (DNA binding domain) of the BRCA2 gene (Figure 2). The OB2 region hosting the identified BRCA2 variant is highly conserved among multiple species. In silico analysis did not report any change in the tertiary structure of the BRCA2 protein. Similarly, the silent mutation was not observed to cause any change in the DNA-protein binding in wildtype and mutated form.
DISCUSSION
In this report, we present a unique case of a male patient with pancreatic and breast cancer with a silent mutation (c.8754 G. A; p.E2918E) in the BRCA2 gene. Over a period of 7 years starting with primary diagnosis of pancreatic cancer, the proband was diagnosed with asynchronous, bilateral, hormone positive breast cancers. Following the NCCN guidelines on genetic testing, the proband was found to be a carrier of a variant c.8754 G>A in exon 21 of the BRCA2 gene, classified as ‘likely pathogenic’ (as per ACMG guidelines). Considering the increased risk of HBOC, prostate cancer and pancreatic cancer in family members, the affected and unaffected family members of the proband were tested for the BRCA2 mutation by Sanger sequencing. Interestingly, two family members of the proband, diagnosed with breast cancer and three unaffected family members were also found to harbour the mutation. Even though deemed as a silent mutation (i.e., without any change in amino acids in BRCA2 polypeptide), this mutation was clinically classified as likely pathogenic and in silico analysis predicted that the mutation may cause aberrant splicing of the BRCA2 gene affecting mRNA stability.
In our report, the association of BRCA2 pathogenic mutation (i.e., genotype) with ER positive status (i.e., phenotype) of male breast cancer is consistent with previous reports (Silvestri et al., 2016). Although not as widely studied as female breast cancers, a few studies have investigated the molecular basis of MBCs (Piscuoglio et al., 2016). Fentiman et. al. 2016 present a comprehensive review of the molecular studies on MBCs and a comparative analysis with female breast cancer cohorts. Luminal A subtype is found to be predominant in MBC with a higher BRCA2 germline mutation frequency than breast cancer in females. Data from the CIMBA cohort analysing the characteristics of BRCA1/2 mutations in MBCs and female breast cancers report pathologic differences (Silvestri et al., 2016). The frequency of BRCA2 mutations in male breast cancer is found to be higher than BRCA1 in comparison to female breast cancers (Silvestri et al., 2016). Recent studies using multigene panel testing also demonstrate differences in the molecular aetiology of MBCs (Moelans et al., 2019; Pritzlaff et al., 2017).
The most interesting finding in our study is the clinically relevant pathogenicity of the c.8754 G>A BRCA2 variant. As a silent mutation, this variant did not induce any amino acid change in the BRCA2 protein and yet, was classified as likely pathogenic in concordance with ACMG guidelines.
Silent mutations have been widely acknowledged to influence changes in protein expression, conformation and function (Sharma et al., 2019). Such mutations have been demonstrated to be capable of altering gene expression through mechanisms such as disruption/creation of splicing regulatory sites, gain/loss of miRNA binding sites and altering mRNA stability. Additionally, silent mutations may also affect changes in translation efficiency (translational kinetics and protein folding) by changing the codons read by tRNAs of different cellular availability (Fernández-Calero et al., 2016). Clinically relevant silent mutations have been reported within the catechol-O-methyltransferase (COMT) gene, an enzyme responsible for degrading catecholamines and a key regulator of pain perception and mood. The p.L136L silent mutation of COMT gene has shown to affect mRNA stability, subsequently having an influence on pain sensitivity and an association with mood disorders including Major depressive disorder (MDD) and bipolar disorder (BD) (Pandolfo et al., 2015). A silent mutation in the DRD2 gene, a dopamine receptor and a key tested candidate gene in psychiatric disorders, results in a less stable secondary structure of the mRNA, further corresponding to the increased mRNA degradation and a lower protein level (Sauna & Kimchi-Sarfaty, 2011). A synonymous mutation (ΔF508) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene has shown to alter the mRNA structure, subsequently leading to a misfolded protein (Chamary & Hurst, 2009). In cancers, such silent mutations in ABCB1 (P-glycoprotein) gene are associated with multidrug resistance via alteration of substrate specificity (Kimchi-Sarfaty et al., 2007). Specific haplotypes of ABCB1 (c.1236C>T, c.2677G>T and c.3435C>T) have been associated with improved treatment outcomes in patients with metastatic renal cell cancer and inflammatory bowel disease (Sauna & Kimchi-Sarfaty, 2011).
Germline BRCA1/2 pathogenic mutations (either point mutations or gene rearrangements) are well established genetic factors associated with predisposition to HBOC. However, the contribution of silent point mutations in these key tumor suppressor genes to HBOC risk remains to be elucidated (Diederichs et al., 2016). Silent mutations in BRCA1 gene have been demonstrated to result in reduced mRNA levels (Findlay et al., 2018). BRCA2 silent mutations have been previously reported to result in exon skipping and changes in protein structure. The study by Hansen et. al. demonstrated that the silent mutation in exon 6 of BRCA2 (c.744 G>A/c.516 G>A, K172K) resulted in exon 6 skipping during splicing. The resulting frameshift causes premature termination of codons 154 or 168 leading to nonsense-mediated mRNA decay, thereby potentially affecting stoichiometric amounts of protein available for biological functions.
In the current study, using a NCCN-guided clinical genetics approach for HBOC familial risk assessment, we have identified a clinically relevant BRCA2 silent mutation (c.8754G>A, p.E2918E associated with male and female hormone positive breast cancer. In the BRCA2 gene, this variant resides in the oligonucleotide binding (OB2) domain. This OB2 region is known to bind to DSS1, an evolutionarily conserved acidic protein, which is linked to stabilizing BRCA2, promoting homologous recombination and implicated in various other processes including development, and protein degradation (Pfeffer et al, 2017). In agreement with our bioinformatics analysis, the c.8754G>A variant leads to an alteration of the canonical donor site, and the splicing outcome leads to a 46-nt insertion of intron 21 resulting in a frameshift and premature appearance of a stop codon (Acedo et al., 2015). This identified variant was reported to induce major aberrant transcripts and thus was classified as “pathogenic” according to the class C5, five-tiered ACMG classification of variants (Acedo et al., 2015; Richards et al., 2015). This mutation and a nearby silent mutation (c.8755) has been reported in Latin American population although the prevalence in other ethnicities population is not well established (Karami & Mehdipour, 2013; Millan Catalan et al., 2019; Rebbeck et al., 2018). Also referred to as the c.8982G>A variant, this variant has been previously reported in individuals afflicted with HBOC syndrome (Finkelman et al., 2012).
Based on our familial risk profiling and in silico analysis of this BRCA2 silent mutation, further functional genomics studies are warranted to understand biological mechanisms that link the genotype of BRCA2 pathogenic variant (i.e, c.8754 G>A) to the HBOC phenotype (i.e, loss of tumor suppressor function). It is likely that such functional studies will reveal newer insights into the BRCA2 biology with focus on mRNA stability associated with homologous recombination processes.
Conclusion
In the current study, we report the presence of a clinically relevant silent BRCA2 mutation (c.8754G.A, p.E2918E) characterised as likely pathogenic in six members of a family, three of whom presented with hereditary breast cancers. Although in silico analysis predicts formation of BRCA2 aberrant transcripts due to this silent mutation, further mechanistic experimental studies will be needed to determine its functional role in carcinogenesis. Analysis of pathogenic silent mutations in key cancer-associated genes are expected to provide newer insights in underlying mechanisms of carcinogenesis.
Data Availability
The study investigators would like to state that all data related to the manuscript will be available for further review on request after consultation with institutional ethics committees
CONSENT
The study was approved by an Independent Ethics Committee. All subjects gave their consent for the use of their personal and medical information associated with this study.
CONFLICT OF INTEREST STATEMENT
The author declares no competing interests.
ETHICS APPROVAL
The study was approved by the Institutional Ethics Committee of PCCM (DCGI/CDSCO Registration Number: ECR/298/Indt/MH/2018. Every participant gave a separate informed consent for the use of their personal and medical information associated with this study. The study follows principles of the declaration of Helsinki (Brazil, 2013 version) and National Ethics Guidelines 2017 issued by the Indian Council of Medical Research, Government of India.
ACKNOWLEDGEMENT
The study authors would like to thank all participants who consented to participate in this study. We acknowledge Bajaj Auto Ltd. for providing support to research activities at Prashanti Cancer Care Mission, Pune. We are thankful to Dr Sanket Nagarkar, Dr Sneha Joshi and Nikki Dutt for their support in preparation of this manuscript.
Footnotes
AUTHOR EMAIL IDS
1. ashwinipccm{at}gmail.com
2. siddharthgahlaut{at}gmail.com
3. rupavr{at}gmail.com
4. aeijazul{at}gmail.com
5. lalehbusheri{at}gmail.com
6. rreddy0811{at}gmail.com
7. shaheens268{at}gmail.com
8. ashraf{at}strandls.com
9. smeetanare{at}hotmail.com
Abbreviations
- BC
- Breast Cancer
- MBC
- Male Breast Cancer
- NCCN
- National Comprehensive Cancer Network
- HBOC
- Hereditary Breast and Ovarian Cancer
- USG
- Ultrasonography
- OB
- Oligonucleotide Binding
- ACMG
- American College of Medical Pathologists guidelines
- NGS
- Next generation sequencing
- gDNA
- Genomic Deoxyribonucleic acid
- ER
- Estrogen receptor
- PR
- Progesterone receptor
- HER2
- Human epidermal growth factor receptor
- CIMBA
- Consortium of Investigators of Modifiers of BRCA1/2
References



