
Summary
Background HIV-1 infections initiated by multiple founder variants are characterised by a higher viral load and a worse clinical prognosis, yet little is known about the routes of exposure through which transmission of multiple founder variants is most likely.
Methods We conducted a systematic review of studies that estimated founder variant multiplicity in HIV-1 infection, searching MEDLINE, EMBASE and Global Health databases for papers published between 1st January 1990 and 14th September 2020 (PROSPERO study CRD42020202672). Leveraging individual patient estimates from these studies, we performed a logistic meta-regression to estimate the probability that an HIV infection is initiated by multiple founder variants. We calculated a pooled estimate using a random effects model, subsequently stratifying this estimate across nine transmission routes in a univariable analysis. We then extended our model to adjust for different study methods in a multivariable analysis, recalculating estimates across the nine transmission routes.
Findings We included 70 publications in our analysis, comprising 1657 individual patients. Our pooled estimate of the probability that an infection is initiated by multiple founder variants was 0·25 (95% CI: 0·21-0·29), with moderate heterogeneity (Q = 132 · 3, p < 0 · 001, I2 = 64 · 2%). Our multivariable analysis uncovered differences in the probability of multiple variant infection by transmission route. Relative to a baseline of male-to-female transmission, the predicted probability for female-to-male multiple variant transmission was significantly lower at 0·13 (95% CI: 0·08-0·20), while the probabilities for people-who-inject-drugs (PWID) and men-who-have-sex-with-men (MSM) transmissions were significantly higher at 0·37 (0·24-0·53) and 0·30 (0·33-0·40), respectively. There was no significant difference in the probability of multiple variant transmission between male-to-female transmission (0·21 (0·14-0·31)), post-partum mother-to-child (0·18 (0·03-0·57)), pre-partum mother-to-child (0·17 (0·08-0·33)), intrapartum mother-to-child (0·27 (0·14-0·40)).
Interpretation We identified PWID and MSM transmissions are significantly more likely to result in an infection initiated by multiple founder variants, whilst female-to-male infections are significantly less likely. Quantifying how the routes of HIV infection impact the transmission of multiple variants allows us to better understand how the evolution and epidemiology of HIV-1 determine clinical outcomes.
Funding This study was supported by the MRC Precision Medicine Doctoral Training Programme (ref: 2259239) and an ERC Starting Grant awarded to KEA (award number 757688). The funding sources played no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Evidence before this study Most HIV-1 infections are initiated by a single, genetically homogeneous founder variant. Infections initiated by multiple founders, however, are associated with a significantly faster decline of CD4+ T cells in untreated individuals, ultimately leading to an earlier onset of AIDS. Through our systematic search of MEDLINE, EMBASE and Global Health databases, we identified 82 studies that classify the founder variant multiplicity of early HIV infections. As these studies vary in the methodology used to calculate the number of founder variants, it is difficult to evaluate the multiplicity of founder variants across routes of exposure.
Added value of this study We estimated the probability that an HIV infection is initiated by multiple founder variants across exposure routes, leveraging individual patient data from 70 of the identified studies. Our multivariable meta-regression adjusted for heterogeneity across study methodology and uncovered differences in the probability that an infection is initiated by multiple founder variants by exposure route. While overall, we estimated that 25% of infections are initiated by multiple founder variants, our analysis found that this probability for female-to-male transmission is significantly lower than for male-to-female transmission. By contrast, this probability was significantly higher among people-who-inject-drugs (PWID) and men-who-have-sex-with-men (MSM). There was no difference in the probability of multiple founder variant transmission for mother-to-child transmission when compared with male-to-female sexual transmission.
Implications of all the available evidence Because HIV-1 infections initiated by multiple founders are associated with a poorer prognosis, determining whether the route of exposure affects the probability with which infections are initiated by multiple variants facilitates an improved understanding of how the evolution and epidemiology of HIV-1 determine clinical progression. Our results identify that PWID and MSM transmissions are significantly more likely to result in an infection initiated by multiple founder variants compared to male-to-female. This reiterates the need for focussed public health programmes that reduce the burden of HIV-1 in these risk groups.
Introduction
Transmission of HIV-1 results in a dramatic reduction in genetic diversity, with a large proportion of infections initiated by a single founder variant.1,2 An appreciable minority of infections, however, appear to be the result of multiple founder variants simultaneously initiating infection after a single exposure.3 Importantly, these infections caused by multiple founder variants are associated with elevated set point viral load and faster CD4+ T lymphocyte decline.4–7
HIV-1 infections initiated via different routes of exposure are subject to different virological, cellular and physiological environments, which likely influence the probability of acquiring infection.8–10 For example, the per-act probability of transmission upon exposure is six times and eighteen times higher for transmission between people who inject drugs (PWID) and men who have sex with men (MSM) than for heterosexual transmission.11
Despite these differences in the probability of HIV-1 acquisition by route of exposure, there is currently no consensus about whether the route of exposure determines the probability that infection is initiated by multiple founder variants. Differences in selection pressure during transmission have been observed between sexual exposure routes, with less selection occurring during sexual transmission from males to females than vice-versa, and less selection during MSM transmission relative to heterosexual exposure overall.12,13 Less selection should lead to more opportunities for infections initiated with more founder variants. Studies quantifying the number of founder variants are, however, inconsistent with these findings, which may be due to differences in methodology and study population.3,12,14,15 Moreover, while acquisition risk during sexual transmission is known to be elevated during conditions that increase mucosal inflammation and compromise mucosal integrity, there is no consistent evidence that PWID transmissions, which bypass mucosal barriers altogether, are associated with a higher probability of founder variant initiation.16,17 To estimate the role of exposure route on the acquisition of multiple HIV-1 founder variants, we conducted a meta-regression leveraging all available individual patient data, and accounting for heterogeneity across methodology and study population.
Methods
Search Strategy and Eligibility Criteria
We searched MEDLINE, EMBASE and Global Health databases for papers published between 1 January 1990 to 14 September 2020 (Appendix S1, ppA2-A6). To be included, studies must have reported original estimates of founder variant multiplicity in people with acute or early HIV-1 infections, be written in English and document ethical approval. Studies were excluded if they did not distinguish between single and multiple founder variants, if they did not detail the methods used, or if the study was conditional on having identified multiple founders. Additionally, studies were excluded if they solely reported data concerning people living with HIV-1 who had known or suspected superinfection, who were documented as having received pre-exposure prophylaxis, or if the transmitting partner was known to be receiving antiretroviral treatment. No restrictions were placed on study design, geographic location, or age of participants. Studies were screened independently by SL and JB. Reviewers were blinded to study authorship during the title and abstract screens, and full text reviews were conducted independently before a consensus was reached; consulting other co-authors when necessary. This review conforms to PRISMA guidelines (Table S2, Appendix ppA7-A10).
Data Extraction
Individual patient data (IPD) were collated from all studies, with authors contacted if these data were not readily available. Studies were excluded from further analysis if IPD could not be obtained. Only individuals for whom a route of exposure was known were included. Additionally, we removed any entries for individuals with known or suspected superinfection, who were receiving pre-exposure prophylaxis or for whom the transmitting partner was known to be receiving antiretroviral therapy. For the base-case dataset, we recorded whether an infection was initiated by one or multiple variants and eight predetermined covariates to be considered in the multivariable meta-regression:
Route of exposure. Female-to-male (HSX-FTM), male-to-female (HSX-MTF), men-who-have-sex-with-men (MSM), pre-partum, intrapartum and post-partum mother to child (MTC), or people who inject drugs (PWID).
Quantification Method. Methodological groupings were defined by the properties of each approach, resulting in six levels: phylogenetic, haplotype, distance, model, or molecular (Table S1).
HIV subtype. Infecting subtypes were classed as either a canonical geographically delimited subtypes (A-D, F-H, J and K), a circulating recombinant form (CRF), or ‘recombinant’ (when a putative recombinant was identified but not designated a CRF).18,19
Delay between infection and sampling. For sexual or PWID exposures, the delay was classified as either less than or equal to 21 days if the patient was seronegative at time of sampling (Feibig stages I-II) or more than 21 days if the patient was seropositive (Fiebig stages III-VI). For mother-to-child infections, if infection was confirmed at birth, or within 21 days of birth, the delay was classified as either less than or equal to 21 days. A positive mRNA or antibody test reported after this period was classified as a delay of greater than 21 days.
Number of genomes analysed per participant. For studies that use single genome amplification, this was the number of consensus genomes obtained.
Genomic region analysed. The region was classified as envelope (env), pol, gag or near full length genome (NFLG).
Alignment length analysed. The length was measured in base pairs, discretised to the nearest 250, 500, 1000, 2000, 4000, 8000, and near full length genome (NFLG) intervals (∼9000).
Use of single genome amplification (SGA) to generate viral sequences. A binary classification was used to characterise whether the viral genomic data were generated using SGA. SGA mitigates the risk of Taq-polymerase mediated template switching, nucleotide misincorporation or unequal amplicons resampling encountered in regular bulk or near endpoint polymerase chain reaction (PCR) amplification.20–22
If information from any of the covariates iii-viii was missing or could not be inferred from the study, we classified its value as unknown. We excluded covariate levels for which there were fewer than 6 data points. For our main analysis, we removed repeat measurements for the same individual, and used only those from the earliest study or, where the results of different methods were reported by the same study, the conclusive method used for each individual. Further details on covariate selection are in the supplementary methods (Appendix ppA2-A6).
Statistical Analysis
We calculated a pooled estimate of the probability of multiple founder variant infection using a ‘one-step’ generalised linear mixed model (GLMM); assuming an exact binomial distribution with a normally distributed random effect on the intercept for within-study clustering and fitted by approximate maximum likelihood.23 Heterogeneity was measured in terms of τ2, the between-study variance; I2, the percentage of variance attributable to study heterogeneity; and Cochran’s Q, an indicator of larger variation between studies than of subjects within studies.24 Publication bias was assessed using funnel plots and Egger’s regression test.25 All analyses were conducted in R 4.1.2.26
Pooled estimates obtained through a ‘one-step’ approach are usually congruent with the canonical ‘two-step’ meta-analysis model, however discrepancies may arise due to differences in weighting schemes, specification of the intercept or estimation of residual variances.27 We compared the results from our ‘one-step’ model with a ‘two-step’ binomial-normal model to confirm our estimates were consistent. We also performed seven sensitivity analyses to test the robustness of our pooled estimate: i) iteratively excluding single studies, ii) excluding studies that contained fewer than ten participants, iii) setting variable thresholds of the number of genomes per patient, iv) excluding studies that consisted solely of single founder infections, v) excluding IPD that did not use single genome amplification, vi) including only those data that matched a ‘gold-standard’ methodology of haplotype-based methods and envelope gene analysis, and vii) an assessment of the effect of vaccine breakthrough, sequencing technologies, and molecular methods. To validate our down-sampling method that used only the most recent study for repeated individual data, we calculated a distribution of pooled estimates by refitting the pooling models to 1000 datasets, each containing one datapoint per individual sampled at random from an individual’s possible measurements.
We extended our ‘one-step’ model by conducting a univariable meta-regression with each covariate contributing a fixed effect and assuming normally distributed random effects of publication. We extended this model to a multivariable analysis. Fixed effects were selected according to a ‘keep it maximal’ principle, in which covariates were only removed to facilitate a non-singular fit and to prevent multicolinearity.28 We defined our reference case as heterosexual male-to-female transmission, and evaluated through a gold-standard methodology of haplotype-based methods, analysis of the envelope genomic region and a sampling delay of less than 21 days. We report stratified model estimates of the proportion of infections initiated by multiple founders and bootstrapped 95% confidence intervals across each covariate with all other covariates held at their reference case values. We performed four sensitivity analyses to test the robustness of the selected multivariable meta-regression model: i) iteratively excluding single studies, ii) excluding studies that contained fewer than ten participants, iii) excluding studies that consisted solely of single founder infections, and iv) excluding IPD that did not use single genome amplification. The re-sampling sensitivity analysis was repeated on our selected multivariable model as described above for the univariable model. Further details are in the supplementary methods (Appendix ppA2-A6).
Role of Funding Source
The funder of the study played no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
Our search found 7416 unique papers, of which 7334 were excluded. Of the remaining 380, 207 were excluded after abstract screening, leaving a total of 82 eligible studies for IPD collation (Fig.1).3,5–7,12,14–17,22,29–102 We extracted IPD from 80 of these studies, comprising 3251 data points. The 80 selected studies from which IPD were collated, were published between 1992 and 2020. Of the 3251 data points extracted, 1484 were excluded from our base case dataset to avoid repeated measurements; arising either between different studies that analysed the same individuals (resulting in the exclusion of five studies), or from repeat analysis of individuals within the same study. After excluding participants for whom the route of exposure was unknown or for whom one or more of their covariate values pertained to a covariate level that did not meet the minimum number (6) of observations across all participants, the base case dataset for our analysis comprised estimates from 1657 unique patients across 70 studies.

PRISMA flowchart outlining our systematic literature search and the application of exclusion criteria for the individual patient data meta-analysis.
Our base case dataset includes a median of 13 participants per study (range 2-124) and represents infections associated with heterosexual transmission (42·0%, (n=696), MSM transmission (37·4%, n=621), MTC (14·1%, n=234), and PWID transmission (6·4%, n=106) (Fig.2; Table 1; Table S2, Appendix ppA11-17). Among heterosexual transmissions, 67·7% (n=471) were HSX:MTF transmissions, 29·9% (n=208) were HSX:FTM transmissions, with the remainder undisclosed (n=17). Similarly, we subdivided MTC transmission according to the timing of infection with 44·4% (n=104) pre-partum, 24·4% (n=57) intrapartum, 4·7% (n=11) post-partum, with the remainder undisclosed (n=62). Our dataset spanned geographical regions and dominant subtypes, capturing the diversity of the HIV epidemic over time (Fig. S1, Appendix ppA17). Across the base case dataset, 37·1% (n=618) estimates used phylogenetic methods, 26·4% (n=438) used haplotype methods, 20·9% (n=347) used molecular methods, and 13·0% (n=215) and 2·35% (n=39) of estimates were inferred using distance and model-based methods respectively (Fig.2).

Individual patient data characteristics from the included studies that were tested for inclusion as fixed effects in the multivariable meta-regression model.
Summary of individual and study characteristics in our base case dataset. Transmission groups recorded as: female-to-male (HSX:FTM), male-to-female (HSX:MTF), men-who-have-sex-with-men (MSM), mother-to-child pre-partum, intrapartum), and post-partum; people who inject drugs (PWID). Numbers within parentheses refer to quantities before removal of repeat participants.
Our binomial GLMM pooled estimated the probability that an infection is initiated by multiple founder variants as 0·25 (95% CI: 0·21-0·29), identifying moderate heterogeneity (Q = 132 · 3, p < 0 · 001, I2 = 64 · 2%). Visual inspection of a funnel plot and a non-significant Egger’s Test (t = - 0·7495, df = 55, p = 0·4568) were consistent with an absence of publication bias (Fig.S9 Appendix ppA26). Sensitivity analyses revealed the pooled estimate was robust to the choice of model, the inclusion of estimates from repeat participants, and to the exclusion of studies that contained fewer than 10 participants (Fig.S2, Appendix ppA19). While restricting the analysis to participants for whom a large (>28) number of sequences were analysed did not change the pooled estimate (0·26 (0·20-0·34)), restricting the analysis to those individuals with fewer than 11 sequences reduced the estimate to 0·21 (0·17-0·25) (Fig.S3 Appendix ppA20). Analysing only data that matched our ‘gold standard’ study methodology slightly increases the pooled estimate (0.28 (95% CI: 0.22-0.35)) (Fig.S3, Appendix ppA20). We did not identify any studies or risk groups that individually influenced the pooled estimate significantly (Fig.S4, Fig.S5 Appendix ppA21-A22). A pooled estimate subgroup analysis of placebo and vaccine participants from studies for which vaccination status was available revealed no discernible influence of trial arm (Fig.S6, Appendix ppA23). Likewise, no discernible difference was identified between sequencing technologies on the pooled estimate (Fig.S7, Appendix ppA24).
We first extended our binomial GLMM with univariable fixed effects. Relative to a reference exposure route of HSX:MTF, we found significantly lower odds of HSX:FTM transmission being initiated by multiple founder variants (Odds Ratio (OR): 0·53 (95% CI 0·33-0·85)), while other exposure routes were not significantly different (Table 2). The univariable analyses also indicated significantly lower odds of identifying multiple founder variants when the near-full-length genome (NFLG) was analysed (OR: 0·38 (95% CI:0·19-0·68)), relative to the envelope genomic region, while molecular methods resulted in significantly greater odds (OR: 1·93 (1·02-3·45)), relative to haplotype methods. NFLG individuals continued to indicate significantly lower odds of identifying multiple founder variants in the absence of individuals analysed using molecular methods (Fig.S8, Appendix ppA25).
Odds ratios that an HIV-1 infection is initiated by multiple founder variants, inferred from fixed effects coefficients from the univariable and multivariable meta-regression model. Significant effects in bold. MSM - men who have sex with men; PWID - people who inject drugs; NFLG - near full length genome.
Next, we used a multivariable model to calculate the probability of multiple founder variants across the seven routes of exposure controlling for method, genomic region, and sampling delay (Fig.3, Table 2). A satisfactory fit was confirmed by inspection of binned residuals superimposed over 95% confidence intervals (Fig. S10, Appendix pp A27). Model estimated probabilities were calculated with respect to our ‘gold standard’ methodology. Compared to a HSX:MTF transmission probability of 0·21 (95% CI: 0·14-0·31), we found that HSX:FTM transmissions were less likely to be initiated by multiple founders than male-to-female transmissions, with probability 0·13 (95% CI: 0·08-0·21) (OR: 0·55 (95% CI 0·34-0·88)). Conversely, PWID and MSM transmissions were more likely to be initiated by multiple founders (0·37 (0·24-0·53) and 0·30 (0·22-0·40), respectively), compared to HSX:MTF (OR: 2·18 (1·11-3·89); 1·61 (1·00-2·34)) (Fig. 3A). Stratifying MTC transmissions by the putative timing of infection, we calculated pre-partum exposures were initiated by multiple founders with probability 0·17 (0·08-0·33), post-partum with probability 0·18 (0·03-0·57), and intrapartum transmissions with probability 0·27 (0·14-0·45).

Model estimated probabilities and coefficients obtained from the multivariable model. A) Model estimated probabilities of an infection being initiated by multiple founder variants, stratified by the route of exposure. B-D) Inferred odds ratios of fixed effects variables. Blue denotes that a covariate level significantly decreases the odds of an infection being initiated by multiple founders, whilst red indicates covariate levels for which the odds are significantly greater. For each plot, the reference case is marked at the top of the y axis (dotted line) and the marker size scales with sample size.
We calculated the accuracy of different methods by comparing their estimated probability of multiple founder variants to a gold-standard methodological reference scenario of haplotype-based methods on whole genome sequences with individuals with less than 21 delays between infection and sampling. Our analysis indicates using model-based methods underestimates the chance of multiple founder variants (OR: 0·36 (95% CI: 0·09-0·87)), while using molecular methods results in an overestimation (OR: 2·05 (1·09-3·53)). Compared to the envelope genomic region, analysis of near-full-length genome fragments likely underestimates the proportion of multiple founder infections (OR: 0·31 (95% CI: 0·13-0·62)). Our sensitivity analyses revealed the odds ratios calculated using the uni-and multivariable models are robust to inclusion of data from repeated participants, and to the exclusion of studies that contained fewer than 10 participants, of studies that consisted solely of single founder infections, and of individual data that did not use single genome amplification (Fig.S11, Appendix ppA28).
Discussion
Using data from 70 published studies, we estimated that a quarter of HIV-1 infections are initiated by multiple founder variants. When controlling for different methodologies across studies, the probability that an infection is initiated by multiple founders decreased from 0·21 (95% CI: 0·14-0·31) for male-to-female infections, to 0·13 (95% CI: 0·08-0·21) for female-to-male infections, but increased for MSM and PWID infections (0·30 (0·22-0·40) and 0·37 (0·24-0·53), respectively). Further, we found that model-based methods, representing a group of approaches that determine founder multiplicity by comparing the observed distribution of diversity with that expected under neutral exponential outgrowth from single variant transmission, were less likely to identify multiple founder infections whereas molecular methods overestimated. Together these results suggest that while the exposure route probably influences the number of founder variants, previous comparison has been difficult due to different study methodologies.
Our pooled estimate is consistent with the seminal study of Keele et al., who found 23·5% (24/102) of their participants had infections initiated by multiple founders.3 Our stratified predicted probabilities, however, were marginally higher than those of previous studies. A nine-study meta-analysis of 354 subjects found 0·34 of PWID infections were initiated by multiple founders compared with 0·37 (0·24-0·53) in our study and 0·25 for MSM infections for which we calculated (0·30 (0·22-0·40) 12 An earlier meta-analysis of five studies and 235 subjects also found PWID infections were at significantly greater odds than heterosexual infections of being initiated by a single founder, with the frequency of founder variant multiplicity increasing 3-fold, while a smaller, non-significant 1·5-fold increase was observed with respect to MSM transmissions.16 In both instances, these studies restricted participants so that the methodology in estimating founder variant multiplicity was consistent. In this study, we were able to leverage individual level data to control for methodological sources of heterogeneity across publications.
Across sexual transmission routes, the probability of multiple founder variants is positively, albeit weakly, associated with an increase in the risk of transmission given exposure. Nonetheless, the probability that infection is initiated by multiple founders remain remarkable consistent. For example, while male-to-male exposures may be up to eighteen times more likely to result in transmission than male-to-female exposures, we calculated a 1·6 fold increase in the risk of multiple founders.11 Previously, Thompson et al. reconciled the low probability of acquisition with the relatively high probability of multiple founders by assuming only a fraction of exposures occur in environments conducive for transmission.103 In sexual transmission, this could be induced through epithelial damage arising from ulceration or microtrauma; enhancing translocation of viral particles or driving inflammation that propagates recruitment of permissive target cells.8 Despite a higher constitutive abundance of permissive cells in the adult human foreskin, the endocervical epithelium and its junction with the ectocervical epithelium are much more susceptible to inflammation and micro-abrasions, reflecting the transmission bias observed in heterosexual transmission.104,105
Our analysis has some limitations. First, our classification of founder variant multiplicity is determined by the individual studies, but explicitly defining a founder variant remains challenging. Recent studies have suggested a continuum of genotypic diversity exists, rather than discrete variants, that gives rise to distinct phylogenetic diversification trajectories and may not be reflected by a binary classification.32,68 Although a threshold is specified for distance-based methods, this often varies between publications.106,107 For example, both Keele et al and Li et al analysed the diversity of the envelope protein, but whilst the former classifies populations with less than 0·47% diversity as homogenous, Li et al included samples up to 0·75%.3,15 The distinction between single and multiple founder variants may further be blurred by recombination and hypermutation.108,109 Our finding that the analysis of near-full-length genomes were associated with a significant decrease in the odds of multiple founders, suggests earlier studies that rely on smaller, highly variable, fragments of envelop, may have overestimated the frequency of infections initiated by multiple founder variants. Similarly, our sensitivity analyses revealed a subtle correlation between the number of genomes analysed and the probability of observing multiple founder variants, pointing to the possibility that using too few genomes could limit the chance of observing multiple founders.
Second, we acknowledge that some heterogeneity associated with our estimates is encapsulated within the classification of route of exposure. Relying on self-reported route of exposure may bias our results if misclassification occurs systematically across studies. Similarly, insufficient data were available to properly consider risk factors such as genital ulceration, early stage of disease in the transmitter or receptive anal intercourse. These risk factors may confound or mediate any association between the exposure type and the probability of multiple founder variants, potentially hindering a deeper mechanistic understanding as to the risk factors underpinning founder variant multiplicity.11 Also, under the hypothesis that the proportion of infections initiated by multiple founders varies by transmission route, our point estimate will be influenced by their relative proportions in our dataset. Globally, it is estimated that 70% of infections are transmitted heterosexually, compared to 42.2% in our dataset.110 Our point estimate should be considered a summary of the published data over the course of the HIV-1 epidemic, and not a global estimate at any fixed point in time.
Finally, for several covariates the bootstrapped confidence intervals are wide and may lead to some uncertainty. These are a product of small sample sizes for certain observations, combined with the random effect of publication used in the meta-regression.
This systematic review and meta-analysis demonstrate that infections initiated by multiple founders account for a quarter of HIV-1 infections across major routes of transmission. We find that transmissions involving PWID and MSM are significantly more likely to be initiated by multiple founder variants, whilst HSX:FTM infections are significantly less likely, relative to HSX:MTF infections. Quantifying how the routes of HIV infection impact the transmission of multiple variants allows us to better understand the evolution, epidemiology and clinical picture of HIV transmission.
Data Availability
Previously published data will be available alongside the code used for this study in a GitHub repository
Contributors
KEA conceived the study. JB, SL, DT, KEA designed the study. JB and SL extracted the data. JB performed the experiments and analysed the data. JB and KEA verified the data. All authors interpreted the data. JB and KEA drafted the manuscript, with critical revisions from all authors. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Declaration of Interests
The authors declare no competing interests.
Data Sharing
Code and individual patient data used in this study is publicly available at https://github.com/J-Baxter/foundervariantsHIV_sysreview.
Acknowledgements
JB was supported by the MRC Precision Medicine Doctoral Training Programme (ref: 2259239); CJV-A and KEA were funded by an ERC Starting Grant (award number 757688) awarded to KEA. We are grateful to Morgane Rolland for agreeing to share additional individual patient data with the authors to complete this study. We thank the four anonymous reviewers for their helpful feedback.
Appendices
Supplementary Methods
Protocol Registration
This systematic review and meta-analysis was registered with PROSPERO following the initial literature search (PROSPERO study CRD42020202672).
Full search query submitted to MEDLINE, EMBASE and Global Health databases
(((((transmi*.af. or found*.af. or bottleneck.af. or single.af. or multiple.af. or multiplicity.af. or breakthrough.ti. or TF.af.) and (virus*.af. or variant*.af. or strain.af. or lineage.af. or phenotyp*.af.)) and (HIV.ti. or HIV-1.ti. or human immunodeficiency virus.ti. or env.ti. or envelope.ti or gag.ti. or pol.ti.)) and ((single genome amplification.af. or sga.af. or sgs.af. or ((sequencing.af. or characterized.af.) and (single genome.af. or deep.af. or whole genome.af. or full length.af. or full-length.af.))) or divers*.af. or distance.af. or poisson-fitter.af. or fitness.af. or (monophyletic.af. or paraphyletic.af. or polyphyletic.af.) or (phylogenetic*.af. and (clade.af. or topology.af. or tree.af. or linked.af. or diver*.af. or distance.af. or sieve.af. or molecular dating.af.)))) not ((SIV.ti,ab. or simian immunodeficiency.ti,ab. or fiv.ti,ab. or feline immunodeficiency virus.ti,ab. or exp Hepacivirus/ or Hepatitis.ti,ab. or exp Flaviviridae/ or Tuberculosis.ti,ab. or Enterovirus.ti,ab. or exp Spumavirus/ or diarrhoea.ti,ab. or diarrhea.ti,ab. or superinfection.ti. or exp Malaria/ or CMV.ti,ab. or HPV.ti,ab. or SHIV.ti,ab. OR exp HIV-2/ or phylogeo*.af. or network.ti. or exp HIV Protease Inhibitors/ or exp HIV Integrase Inhibitors/)))
Databases Queried:
Ovid MEDLINE(R) and Epub Ahead of Print, In-Process & Other Non-Indexed Citations, Daily and Versions(R)
Global Health 1910 to 2020 Week 36
EMBASE & EMBASE Classic 1947 – Sep 11
Data Extraction
Route of exposure
We used the route of exposure of horizontally transmitted infections as reported by the original studies. These data are typically ascertained from risk behaviour questionnaires or enrolment criteria for a study cohort. In the majority of cases, a single route of exposure was reported. We stratified the route of exposure as much as possible, given the data available. This resulted in the following levels being included in the models: Female-to-male (HSX-FTM), male-to-female (HSX-MTF), men-who-have-sex-with-men (MSM), pre-partum, intrapartum and post-partum mother to child (MTC), and people who inject drugs (PWID)).
We refer to these as routes of exposure, rather than transmission route as an element of uncertainty is always present; both because there can be multiple concurrent routes of exposure and more generally because self-reported exposure does not necessarily match with transmission. For the same reason we chose not to stratify sexual exposure route into receptive vs. insertive anal sex for male-to-male and vaginal vs. anal sex for male-to-female.
We categorised mother-to-child (MTC) infections into pre-partum, intrapartum and post-partum was using the criteria below. Where these data were nor reported or the results ambiguous, we used the category ‘unknown timing’ (Bertolli et al., 1996):
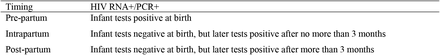
Sampling Delay
For horizontally transmitted infections, the delay between infection and sampling (not diagnosis) was determined according to seropositivity. A delay of less than or equal to 21 days was recorded if the patient was seronegative at time of sampling (Feibig stages I-II) or more than 21 days if the patient was seropositive (Fiebig stages III-VI). For vertical transmissions, if infection was confirmed at birth, or within 21 days of birth, the delay was classified as either less than or equal to 21 days. A positive mRNA or antibody test definitively reported after this period was classified as a delay of greater than 21 days.
Methodologies
A binary classification (yes or no) was inferred as to whether the viral genomic data were generated using SGA. Regular bulk or near endpoint polymerase chain reaction (PCR) amplification can generate significant errors such as Taq-polymerase mediated template switching, nucleotide misincorporation or unequal amplicons resampling. (Meyerhans et al. 1990, Simmonds et al. 1990). In SGA, serial dilutions of viral nucleic acids are made, which, assuming the proportion of positive PCR reaction at each dilution follows a null Poisson distribution, reduces the final reactions to contain a single variant that can be cloned, sequenced, and then analysed. (Simmonds et al. 1990, Salazar-Gonzalez et al. 2008).
Quantification method groupings were defined by the properties of each approach, resulting in six levels: phylogenetic, haplotype, distance, model, or molecular (Table S1). Prior the widespread application of sequencing, molecular methods such as heteroduplex mobility assays could provide a qualitative measure of diversity (Novitsky et al. 1996). Heterogeneous genomic segments would form heteroduplexes during gel electrophoresis of viral RNA, allowing one to distinguish genetically similar and dissimilar segments. Although estimates derived from these assays were regarded as close approximations of viral diversity, they only consider a tiny fraction of the whole genome and cannot provide further information regarding phylogenetics, or functional attributes of any substitutions. As a result they may lead to overestimation of the number of founder variants initiating infection.
Distance and model-based methods assume a threshold or distribution of diversity that is reasonably expected to occur under a hypothesis of neutral exponential growth from a single founder and determine whether the observed diversity is consistent with the modelled values (Slatkin and Hudson, 1991; Lee et al., 2009). Within the model category, we include any mathematical or statistical model which tests whether the observed patterns of diversity can be explained by the transmission of a single variant. For example, this includes Poissonfitter, where frequency distributions of Hamming Distances that significantly diverge from the expected Poisson distribution, after controlling for APOBEC mediated hypermutation, represent an over-dispersed population (Giorgi et al., 2010); simple probabilistic models expressing the expected number of substitutions, and estimates of the time to most recent common ancestor that do not involve the reconstruction of genealogies.
Groupings of methods used to infer the founder variant multiplicity of HIV-1 infections. Model and phylogenetic methods may present as similar metrics such as the most recent common ancestor (tMRCA) and topology, but model-based approaches, unlike phylogenetic methods, do not use genealogical information in their calculation and instead are statistical models applied directly to the genomic data.
Haplotype methods identify linkage patterns of individual polymorphisms across samples from a patient. In the study of HIV founder infection multiplicity, this category mostly concerns the use of highlighter plots, that visually map nucleotide mismatches along an aligned gene segment (Keele et al. 2008). Inspection of these graphs facilitates an approximate enumeration of the number of variants initiating an infection and allow for inference of putative recombinants and APOBEC mediated hypermutation, which would erroneously inflate diversity measures. Haplotype methods may also refer to modelling the distribution of haplotypes obtained through longitudinal deep-sequence samples.
Phylogenetic methods are here defined as approaches that explicitly reconstruct ancestral genealogical relationships directly from sequence data. These either use recipient sequences only, in which case a star-like topology is expected to be observed for single founder infections or use source and recipient sequences from known transmission pairs, such that the number of distinct clades of recipient sequences nested within the source sequences corresponds to the number of founder variants.
Statistical Models
Pooled estimates models
We assumed a binary outcome yik of whether the infection of individual k of study i was initiated by multiple founder variants (1) or not (0) with probability (yik For the two-step model, we first fit a logit model to these binary outcome data for each study, i, where θi is the effect size of study i, xik is whether the infection of individual k of study i was initiated by multiple founder variants (1) or not (0) and ai is the intercept :
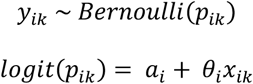
We then accounted for between study variation in the effect sizes by assuming a random effects model such that each of the estimated study effect sizes, 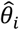 , is a sample from a different normal distribution with a mean equal to an underlying study-specific effect size. This study-specific effect size is itself drawn from a normal distribution with constant mean and variance, τ2 (the between-study variance) :
, is a sample from a different normal distribution with a mean equal to an underlying study-specific effect size. This study-specific effect size is itself drawn from a normal distribution with constant mean and variance, τ2 (the between-study variance) :
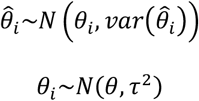
For the one-step model, individual-level and study-level variation are considered simultaneously:
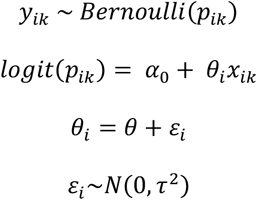
Here, α0 represents a fixed intercept and θi is the random effect of study acting on observation xik. θi is the sum of θ, the mean study effect size, and εi, the study-specific effect drawn from a normal distribution with variance, τ2 (the between-study variance). We compared the results from our one-step model with a two-step model to confirm our estimates were consistent.
Univariable and multivariable models
We extended our one-step model by conducting a univariable meta-regression with each covariate contributing a fixed effect and assuming normally distributed random effects of publication. We report results for univariable models that analysed the role of route of exposure, quantification method, genome region analysed and sampling delay as fixed effects (βik).
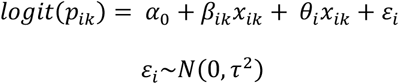
A multivariable model was built from the fixed effects used in the univariable analysis. The fixed effects (βnik, n ∈ [1, m]) were selected according to a ‘keep it maximal’ principle, in which covariates were only removed to facilitate a non-singular fit. The selected model is outlined here:
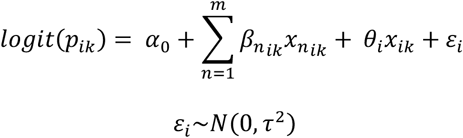
The selected model was assessed for convergence, singularity, multicollinearity using the R package ggeffects. We calculated the proportion of binned residuals within 95% confidence limits. Model estimated probabilities per transmission route were calculated controlling baseline covariates as our ‘gold standard’ methodology (envelope genomic region, a short delay, haplotype analysis).
Software and Computational Methods
All code associated with this study is available under GNU General Public License v3.0 at the following GitHub repository: foundervariantsHIV_sysreview. Further details on how to run the analysis are included in the README.md.
The analyses were conducted in R 4.1.2, principally using the following packages:
lme4, 1.1-27.1, (Bates et al. 2007)
metafor, 3.0-2, (Viechtbauer 2010)
tidyverse, 1.3.1, (including ggplot2 3.3.5, stringr 1.4.0, forcats 0.5.1 & dplyr 1.0.7) (Wickham et al., 2019)
reshape2 1.4.4 (Wickham, 2012)
ggeffects 1.1.1 (Lüdecke, 2018)
mltools 0.5.2
parallel 3.6.2
Prisma Checklist
PRISMA checklist referencing the necessary steps taken to pages in this manuscript.
Table of Selected Studies
Included studies selected for inclusion from our systematic literature search. We record the route of transmission: female-to-male (HSX:FTM), male-to-female (HSX:MTF), men-who-have-sex-with-men (MSM), mother-to-child pre-partum (MTC:PreP), intrapartum (MTC:IntP) and post-partum (MTC:PostP); people who inject drugs (PWID), or nosocomial (NOSO). Additionally, we tabulate the method grouping used to infer founder multiplicity, the genomic region analysed, the number of participants analysed, and the proportion of infections initiated by multiple founders reported by each study. We note the number of single and multiple founder infections included within our base case dataset
Temporal Structure of Exposure and Method

Distributions of transmission route (A), grouped method (B) and sequencing technology (C) over time, highlighting the epidemiologic and methodological step-changes that occurred over the three decades in which the selected studies were published. This means that earlier methods may be biased to those transmission routes that were more common in earlier studies.
Sensitivity Analyses of Pooling

A comparison of the pooled estimates of the probability that an infection is initiated by multiple founders by the one-step (GLMM) and two-step (Binomial-Normal (B-N)) models and respective sensitivity analyses. Plot (A) shows both models calculate concordant estimates and are robust to sensitivity analyses designed to test our inclusion/exclusion criteria, and biases introduced by small or minimal-effect studies. B) reports the distribution of estimates, recalculated from 1000 datasets in which the representative datapoint for each individual was sampled at random from a pool of their possible measurements. The dashed lines and shaded areas denote the original point estimate and confidence intervals, respectively.
Influence of Methodology and Number of Genomes Analysed
We analysed data from participants spanning the interquartile range (11 - 28 genomes), and then restricted the analysis to participants with higher than the upper quartile value (numbers of genomes>28) or lower than the lower quartile value (number of genomes <11). Restricting the analysis to participants for whom a large (>28) or small (<11) number of sequences were analysed adjusted the pooled estimate to 0·26 (0·20-0·34) and 0·21 (0·17-0·25), respectively (Fig.S3). The model fitted to participants spanning the interquartile range also revealed a slight increase in the probability of observing multiple founder variants when compared to the original estimates (0·27 (0·24-0·31)). These findings suggest the presence of a subtle correlation between the number of genomes analysed and the probability of observing multiple founder variants.

Comparing the pooled estimates of the probability that an infection is initiated by multiple founders by the one-step (GLMM) and two-step (Binomial-Normal (B-N)) models under our ‘gold-standard’ methodology, and when varying the threshold of the number of genomes analysed per patient.
Leave-One-Out Cross Validation: Studies

For both one-step and two-step models, we visually inspect the influence of each study included in our analysis on the pooled estimate that an infection is initiated by multiple founders. We find that in iteratively excluding individual studies, no discernible impact on the overall pooled estimate is made. The dashed lines and shaded areas denote the original point estimate and confidence intervals, respectively.
Leave-One-Out Cross Validation: Transmission Routes

For both one-step and two-step models, we visually inspect the influence of each risk group included in our analysis on the pooled estimate that an infection is initiated by multiple founders. We find no discernible impact on the overall pooled estimate is made. The dashed lines and shaded areas denote the original point estimate and confidence intervals, respectively.
Comparison of Vaccine Escape and Placebo Participants
Some of the selected studies included participants enrolled on vaccine trials. As breakthrough infections of vaccine-recipients may not reflect natural infection, we compare vaccine and placebo arms of trials for which these data were available. This analysis included participants from HTVN502 and RV144 (A third vaccine trial (HTVN505) is not included as participant vaccine status was not available). Estimates of founder multiplicity were extracted from Rolland et al 2011 (HTVN502), and Janes et al 2015 (RV144), following our inclusion criteria of selecting the first instance for which data are available (HTVN502 participants were also subsequently analysed by Janes et al.). We did not find any significant difference between vaccine-breakthrough and placebo infections.

A) The proportion of infections identified as being initiated by multiple founders, segregated by vaccine status. B) Pooling estimates calculated using one and two-step models for vaccine trial only datapoints, compared to the base case dataset. C) Univariable analysis finding no significant difference in the odds of observing multiple founder variants between vaccinated and placebo arms of vaccine trial participants.
Comparison of Sequencing Technologies
Of 70 selected studies in our base case dataset, eleven studies used deep sequencing (Roche 454 – 9 studies, Ilumina – 1 study, PacBio HiFi-1 study). To investigate whether the higher resolution of deep sequencing approaches influenced the observation of multiple founder variants initiating HIV infection, we conducted a univariable regression across those studies that used sequence-based methods. (0.22 (95% CI: 0.19-0.27)) was slightly lower than our original pooled estimate (0·25 (95% CI: 0·21-0·29)). In our univariable analysis, we did not find the odds of observing multiple founder variants differed significantly across sequencing methodologies.

A) The proportion of infections identified as being initiated by multiple founders, segregated by sequencing technology. B) Pooling estimates calculated using one and two-step models for sequence methods only datapoints, compared to the base case dataset. C) Univariable analysis finding no significant difference in the odds of observing multiple founder variants across sequencing technologies.
Evaluating the Impact of Molecular Methods
Both our multivariable and univariable analyses identified differences in the probability of multiple founders according to the genomic region analysed. Molecular methods, which we defined as approaches that rely on the formation of heteroduplexes during gel electrophoresis of viral RNA, are very sensitive. This allows one to distinguish genetically similar and dissimilar segments, however the use of these methods on short fragments of envelope may produce false positive results. Indeed, in both our univariable and multivariable analyses, there were significantly greater odds of recording multiple founder infections if molecular methods were used. To evaluate the impact of molecular methods as a confounder on the genomic region, we recalculated our pooled estimate under different scenarios and re-fitted a univariable model of genomic region in the absence of molecular methods. Of 1657 individuals, 1315 in our base case dataset were analysed using the envelope genomic region. Pooled estimates for envelope only individuals and envelope only individuals without molecular methods under the GLMM were 0·28 (0·23-0·32) and (0·25 0·21-0·29) respectively. A univariable analysis of genomic region fitted to the main dataset excluding molecular methods reported findings consistent with the main univariable analysis.

A) The founder variant multiplicity of 1315 individuals was analysed using the envelope genomic region, here segregated by method and indicating the prevalence of multiple founder infections. B) Pooled estimates calculated using one and two-step models from individuals for whom the envelope genomic region was analysed including/excluding molecular methods. C) Univariable analysis on dataset excluding molecular methods, reporting findings consistent with the main univariable analysis.
Evaluation of Publication Bias

Funnel plot to visually evaluate the presence of publication bias. In the absence of publication bias, study estimates are distributed symmetrically with respect to the pooled estimate (vertical solid black line). Here, the log odds of an infection being initiated by multiple founders for each study, plotted against the standard error for each study indicate an absence of publication bias. This conclusion was supported by an Egger’s Regression Test: t = - 0·7495, df = 55, p = 0·4568.
Binned Residuals Plot

Binned residuals from the select multivariable model. 97% of the average residuals across each bin fall within the 95% confidence intervals (white area), indicating a good model fit.
Sensitivity Analyses for Meta-regression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Odds ratios that an infection is initiated by multiple founders, stratified by route of transmission, as calculated in the main analysis (A), following the iterative exclusion of individual studies (B) and bootstrapped estimates recalculated from 1000 datasets in which the representative datapoint for each individual was sampled at random from a pool of their possible measurements (C). Panel (D) plots the odds ratios of all covariate levels included in the meta-regression, stratifying by previously defined sensitivity analyses. Overly generous confidence intervals in (D), particularly under the condition of single genome analysis (SGA) only data, is likely due to small sample sizes in at those levels (n<10).
Footnotes
We have made several important updates: First, we have conducted additional sensitivity analyses investigating the impact of sequencing technology, vaccination, and the number of samples, on the probability that infection is initiated by multiple founders. Second, we have updated our analysis to incorporate a reconsidered selection of "gold standard" or "reference" methodology. We reflected on the issue that in most cases the genomic envelope regions is used to assess founder variant multiplicity, and that for practical reasons it may not always be possible to extract whole genome sequences. Therefore, we determined it was appropriate that our analysis should mirror best practice as currently deployed. Finally, we have updated our analysis to remove a couple of instances of participant duplication. We excluded seven participants, leaving our base case dataset comprising 1657 participants from 70 studies. This update did not change our pooled estimate.
References
Supplementary References



