
ABSTRACT
Background The underlying pathogenetic factors generating the innate immune signal necessary for T cell activation, initiation and chronification of Hidradenitis suppurativa (HS, also known as Acne inversa) are still poorly understood. Emerging evidence suggests that defective keratinocyte function critically contributes to HS disease development and progression.
Objectives To elucidate the role of keratinocytes in HS lesion formation, we compared the transcriptomes of isolated lesional and perilesional HS epidermis by RNA sequencing.
Methods Lesional and perilesional HS skin samples of at least 3 different donors were obtained. Isolated epidermal keratinocytes were further processed for cell culture, protein extraction, immunostaining procedures or RNA isolation and RNA sequencing. For large scale promotor site analysis, DEGs were analyzed for overrepresented transcription factor binding sites. Functional annotation clustering for analyzing enriched functional-related gene groups was performed employing the DAVID Bioinformatics Resources.
Results We show that HS is characterized by a strong epidermal stress state as evident by a significant overrepresentation of an AP-1-driven stress signature in the overall gene expression pattern of lesional keratinocytes and a substantial activation of the stress-activated cJun N-terminal kinase (JNK) pathway in lesional HS epidermis. Additionally, our data reveal a strong induction of STAT1 activation in lesional HS epidermis that likely results from IFNγ production and governs the expression of key inflammatory genes that coordinate activation of innate immunity and the adaptive T cell response in HS.
Conclusions Taken together, these data implicate a new role of combined stress signaling and JAK/STAT1 pathway activation in disease progression of HS suggesting interference with JAK/STAT1 signaling as a potentially promising therapeutic approach for HS.
INTRODUCTION
Hidradenitis suppurativa (HS, also known as Acne inversa) 1-3 is a chronic inflammatory skin disease characterized by recurrent deep-seated and painfully inflamed lesions in the intertriginous apocrine gland-bearing areas of the body 4. Both a T cell–dependent adaptive response and innate immune activation play key roles in disease progression. Those two processes are tightly interwoven since activation of the innate immune system is a prerequisite for the generation of T cells and promotes further adaptive immune responses 5. In HS, both Th17 cell activation 6, regulatory T cell (Treg) deficiency 7, and a strong non-specific inflammation 3,8 correlate with disease progression. However, the underlying pathogenetic factors of HS generating the seminal innate immune signal that bridges T cell activation, initiation and chronification of the disease remain to be elucidated.
There is accumulating evidence that keratinocyte dysfunction plays an important role in HS. For instance, a functional defect of hair follicle keratinocytes 9 and imbalances in epithelial cell differentiation 3 were proposed to sensitize for inflammatory responses thereby contributing to HS evolution. Additionally, stimuli implicated to promote disease persistence involve bacterial overgrowth 9,10 and release of ruptured hair follicle contents (including bacteria, damage-associated molecular patterns (DAMPs), keratin fibers and sebum) into the surrounding tissue 1. Both signals stimulate other cells, including keratinocytes to produce inflammatory cytokines 11. Moreover, we demonstrated a role of epidermal inflammasome activation in the pathogenesis of HS 12. The inflammasome complex controls generation of active interleukin (IL)-1β, which is a critical regulator of inflammation and driver of antigen-specific adaptive immune responses 13. In HS, enhanced IL-1β production from both epidermal keratinocytes 12 and keratinocytes isolated from hair follicles 9 has been reported. These data raise the possibility that increased inflammation during disease progression may critically depend on dysregulation of keratinocyte-dependent gene responses fostering a vicious cycle of proinflammatory events that culminate in persistent chronic inflammation in HS.
To gain better insight into the exact contribution of keratinocytes to lesional gene expression, we performed RNA sequencing of lesional versus perilesional HS epidermis. Unlike previous whole-skin proteome, transcriptome, exome or mRNA microarray analyses 14-19, which did not distinguish between keratinocytic and non-keratinocytic responses, our study provides a first comprehensive overview of the isolated transcriptional pattern of lesional epidermal keratinocytes without major obscuring effects of other immunologically active cells. Our data identify a complex epidermal stress response to external and internal stimuli that is characterized by a significant overrepresentation of an AP-1-driven stress signature in the overall gene expression pattern of lesional keratinocytes. Additionally, we observe a strong induction of STAT1 activation in lesional HS epidermis that likely results from IFNγ production and governs the expression of key inflammatory genes orchestrating activation of innate immunity and the adaptive T cell response. Our data implicate a new role of combined stress signaling and JAK/STAT1 pathway activation in disease progression of HS.
MATERIALS AND METHODS
Isolation of HS skin samples and epidermal sheets
Human skin samples were obtained by in-house therapeutic skin surgery from HS patients after informed consent (ethical vote was given by the local Ethics Committee, University of Würzburg, Bavaria, Germany; No. 306/12). Skin samples of at least 3 different donors were used for experiments. Lesional and perilesional (excised at 2 cm distance from the next active HS lesion) HS skin biopsies of 8 mm in diameter were taken and epidermis and dermis separated by dispase treatment and subsequent mechanical separation as described 20. After trypsinization isolated epidermal keratinocytes were further processed for cell culture, protein extraction or RNA isolation.
RNA-sequencing and bioinformatic analysis
mRNA was extracted from five pairwise-matched lesional and perilesional epidermal HS pellets using the Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and RNA sequencing was performed as detailed in the supplementary informations. For large scale promotor site analysis, DEGs were analyzed for overrepresented transcription factor binding sites represented in the Jasper 2018 database using Pscan (v1.4 21). Functional annotation clustering for analyzing enriched functional-related gene groups was performed employing the Data base for Annotation, Visualization, and Integrated Discovery (DAVID Bioinformatics Resources) (v6.8; david.abcc.ncifcrf.gov) as described 22.
Cell culture
Human epithelial keratinocytes (hEK) isolated from lesional epidermal HS sheets were expanded and cultured as previously described 23. Cells were stimulated with tumor necrosis factor (TNF)α (50 ng/ml), type I interferon (IFN) (10 U/ml; both from R&D Systems, Wiesbaden, Germany) or IFNy (100 ng/ml; Peprotech, Hamburg, Germany) by diluting them directly from appropriate stock solutions into the medium. The selective JAK1 inhibitor upadacitinib (250nM; #S8162; Selleckchem, Munich, Germany) was preincubated for 30 min at 37°C prior to addition of the stimuli.
Immunoblotting analysis of secreted proteins
Cell pellets from lesional and perilesional HS epidermis were lysed and protein expression analyzed by immunoblot as described before (Frings et al., 2019) using the following primary antibodies: p-SAPK/JNK (#9251) and p-STAT1 (#9167; both from Cell Signaling Technology, Frankfurt, Germany), Anti-α-tubulin (#T5168, Sigma-Aldrich, Darmstadt, Germany). Proteins were visualized by enhanced chemiluminescence using appropriate horseradish-peroxidase (HRP)-coupled secondary antibodies.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA isolation, cDNA synthesis and qRT-PCR of lesional and perilesional epidermal HS cell isolates was performed as described previously 20 using the SYBR Green method and appropriate gene-specific primers (Sigma-Aldrich; Table S1). Expression of TLR4 and Keratin 14 was investigated to ensure purity of isolated epidermal sheets 20. Test gene expression was normalized to the expression of the housekeeping gene GAPDH (Table S1) and related to an experimental control using the ΔΔCT method.
Immunostaining procedures
3 μm-skin sections from formalin-fixed and paraffin-embedded biopsies from healthy donor skin (excess tissue after tumor surgery) or perilesional and lesional areas from skin samples of HS patients were selected for immunohistological analysis and stained for p-SAPK/JNK (#4668, Cell Signaling), p-c-Jun (#3270, Cell Signaling), p-STAT1 (#9167, Cell Signaling), and c-Jun (#ab218576, Abcam, Cambridge, UK) proteins using antibodies from Cell Signaling Technology. Antibodies were used at 1:100 (pJNK, pJun), 1:200 (c-Jun) or 1:500 (p-STAT1) dilution and visualized using a HRP system (#K5003; Dako, Hamburg, Germany) 12.
Statistical analysis
If not indicated otherwise, data are depicted as averages ± s.d. from at least three independent experiments. Statistical significance of differences was determined by paired t-test or one-way analysis of variance (ANOVA) with post-hoc Dunnett correction and considered significant at p ≤0.05 (*). Higher significances are indicated accordingly (**≤0.01, ***≤0.001). Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad software Inc., La Jolla, CA).
RESULTS
HS epidermis is characterized by a complex stress response that may influence innate and adaptive immune activation
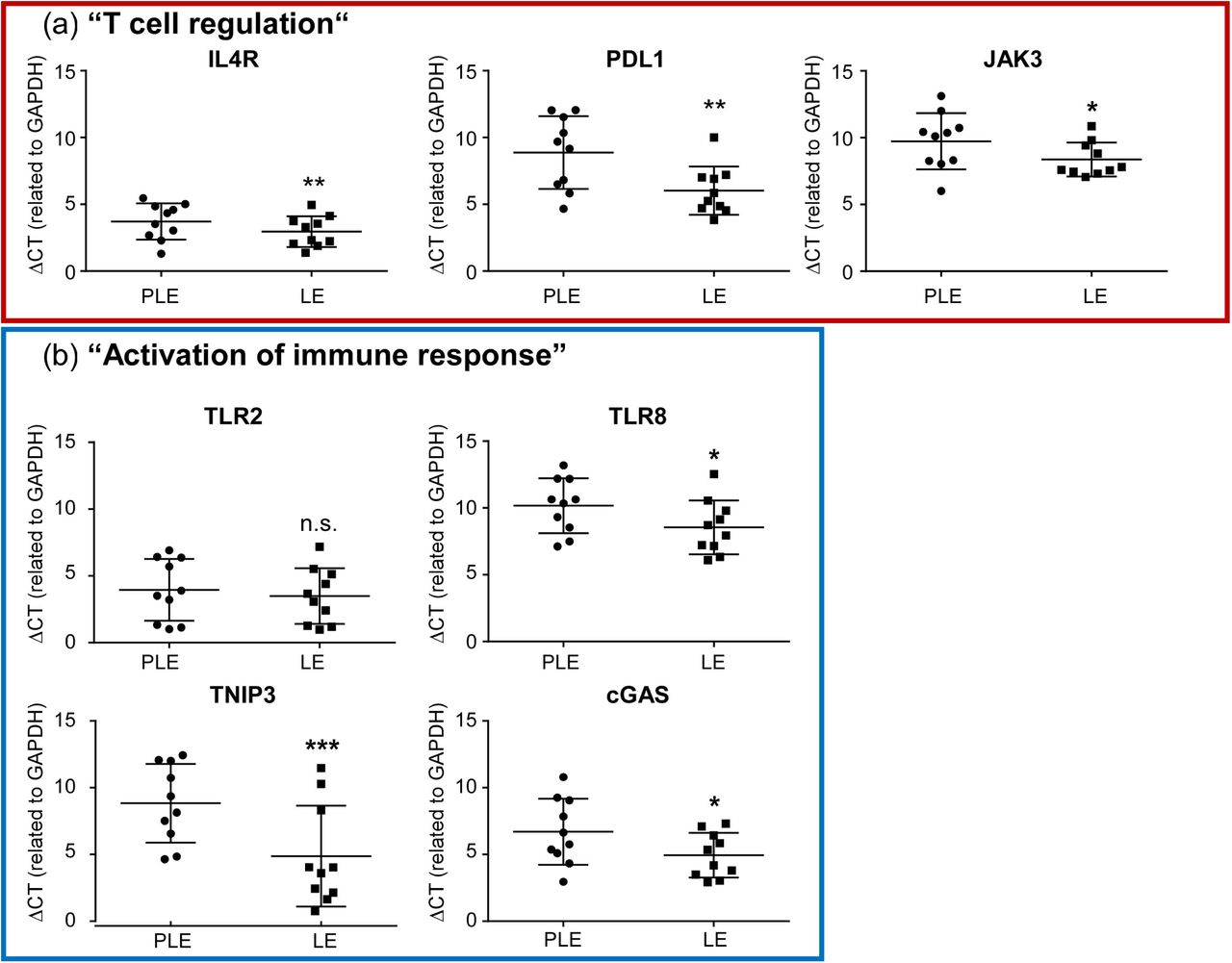
To gain insight into the repertoire of in situ expressed keratinocyte genes in HS we studied pairwise matched perilesional and lesional epidermis isolated from tissue after HS surgery by RNA sequencing. Principal component analysis (Figure S1) showed clustering of expression profiles across patients but clear differences between the transcriptomes of lesional and perilesional HS epidermis. This was unlikely to result from incomplete isolation of basal keratinocytes since both qRT-PCR and sequencing analysis consistently verified high expression of the basal keratinocyte marker K14 and lack of Toll-like receptor (TLR)-4 (Table S2). Expression of the latter is prominent in dermal fibroblasts and macrophages 24 but absent in epidermal keratinocytes 20. In addition, there was no major contamination with non-keratinocytic immune cells since sequencingcounts for immunological markers such as CD3 for T-cells, CD19 for B-cells, CD68 for macrophages, or MPO for neutrophils were extremely low and expression ranged close to the detection limit (Table S2). By statistical evaluation we totally identified 183 up- and 49 downregulated genes in lesional HS epidermis with significantly altered mRNA abundance of ≥ 2-fold (full dataset publicly available at EGA European Genome-Phenome Archive, https://ega-archive.org, ID EGAS00001005544). We next performed functional annotation cluster (FAC) analysis on the differentially expressed genes (DEGs) to identify statistically enriched groups of lesionally regulated functional genes (Figure 1). Consistent with previous reports demonstrating disturbancies of the cutaneous microbiome and a role of neutrophils in HS 12,25-29, we observed a significant overrepresentation of various gene ontology (GO) groups related to cellular responses to bacterial products and neutrophil chemotaxis among the top-upregulated FACs. Additionally, we found enrichment of several GO groups related to leukocyte differentiation, T cell activation, cytokine signaling and innate immune activation. To confirm our findings by an alternative method we performed qRT-PCR for various statistically upregulated gene products from selected functional groups overrepresented in the HS lesions (Figure 2). Since perifollicular deficiencies in Treg numbers or function and imbalance of the Th17/Treg ratio 7 appear to play a central role in HS inflammation, we focused our analysis on genes of the functional cluster “T cell regulation” (IL4R, PDL1, JAK3; Table S3). Additionally, we chose representative genes of the cluster “activation of immune response” (TLR2, TLR8, TNIP3, cGAS; Table S3) containing several genes regulating innate immunity since an altered expression of innate immunity markers may explain the development of chronic inflammatory lesions 2 or contribute to initiation of the dominant T-cell immune response in HS 1. qRT-PCR confirmed significant lesional induction of all tested DEGs (IL4R, PDL1, JAK3, TLR2, TLR8, TNIP3, cGAS; Figure 2) except of TLR2, which expression in lesional skin was increased without reaching statistical significance.

Table with all statistically enriched FACs as obtained using DAVID FAC analysis (v6.8; https://david.abcc.ncifcrf.gov; 54) with mean collective p-values of the included GO groups ≤ 0.01 (as calculated by Fisher’s exact test). Designated functional clusters are ranked according to their enrichment score defined as -log of the collective mean p-value of the included GO groups. The top ten upregulated FACs are presented in bold script, the further analyzed gene clusters “T cell regulation” and “Activation of immune response” are highlighted in red and blue respectively. GO group= gene ontology group.

Comparative qRT-PCR analysis of mRNA expression of selected DEGs from the overrepresented FACs “T cell regulation” (a, framed in red) and “activation of immune response” (b, framed in blue) in lesional vs. perilesional epidermis of HS patients. Data represent mean ΔCT values ± s.d. determined for the respective gene in relation to GAPDH expression. Asterisks indicate statistically significant mRNA induction in n=10 experiments (*p≤0.05, **p≤0.01, ***p≤0.001 paired t-test; CT values for the indicated gene <40 cycles). CT, cycle threshold; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ns, not significant; PLE, perilesional epidermis; LE, lesional epidermis.
Epidermal HS lesions show prominently enriched AP-1- and STAT1-binding sites in the promoters of upregulated genes
To gain better insight into the regulatory pathways underlying the observed transcriptional changes in lesional HS epidermis, we next performed large scale promoter analysis of all ≥2.0-fold statistically DEGs (Table S4). These studies revealed a prominent enrichment of promoter binding sites for multiple AP-1 transcription factors and members of the JAK-STAT signaling pathway among the lesionally upregulated DEGs (Figure 3). DEGs with well conserved AP-1 sites in their promoters involved several of the top upregulated genes such as the antibacterial defensin genes DEFB4B and DEFB4A, the A20-binding protein TNIP3 or the neutrophil-attracting chemokine CXCL8 (Table S5). These genes also consistently appear in the top-regulated FACs (Table S3). Surprisingly, binding sites for NF-κB transcription factors were less prominent among the lesionally induced genes. In particular, we failed to detect statistical overrepresentation of the NF-κB transcription factor cREL (Table S4), which commonly mediates expression of key proinflammatory genes in response to stimulation with HS-relevant cytokines such as TNF and IL-1β 30. Moreover, binding sites for NF κB subunits NFKB1 or NFKB2 were not statistically overrepresented and Rel B sites were only enriched in the lesionally downregulated DEGs (Table S4 and Figure 3). Only binding sites for Rel A showed statistical enrichment in the promoters of the upregulated DEGs albeit with a >500 fold lower enrichment score (ES: 114.81, equivalent to p≤ 8.7E-3; Table S4) than e.g. STAT1 (ES: 61784.46, p≤ 1.6E-5) or the AP-1 factor FOSL1 (ES: 784473.70, p< 1.3E-6), suggesting a more prominent contribution of AP-1 factors and JAK/STAT signaling to the keratinocytic gene expression profile in HS lesions.

Overrepresented TF binding sites within the promoters of all ≥ 2.0 fold differentially regulated epidermal genes in lesional vs. perilesional HS samples as identified by large scale promoter analysis (PSCAN; 21). Blue colored bars represent overrepresented AP-1 TF sites, red colored ones sites corresponding to the JAK-STAT signaling pathway and purple ones TF sites of the NF-κB signaling pathway. Grey bars denote overrepresented sites belonging to other TF families. The overrepresentation score corresponds to 1/p-value. Cut-off for upregulated TF binding sites was arbitrarily set at an overrepresentation score >1000.00 (corresponding to p <0.001), for downregulated ones at >20 (corresponding to p-value <0.05). TF, transcription factor.
Activation of AP-1 factors is a well-known consequence of cytokine- and microbially induced stress signaling and is usually triggered via phosphorylation by stress-activated protein kinases such as c-Jun N-terminal kinase (JNK) 31. Additionally, AP-1 activation is regulated at the transcriptional level e.g. by activation of the mitogen-activated protein kinases ERK1/2 32. Initial analysis of our RNA sequencing data revealed no overt differences in the expression of individual AP-1 factors in lesional vs. perilesional HS epidermis (data not shown). However, we observed a strongly increased epidermal phosphorylation of c-Jun and JNK in lesional HS epidermis (Figure 4), suggesting that enhanced JNK-mediated stress signaling may account for the observed overrepresentation of AP-1 sites in the regulated transcriptome of lesional HS epidermis.

(a) Immunohistochemistry shows an increased JNK and c-Jun phosphorylation in lesional as compared to intraindividual perilesional HS skin. Healthy donor skin served as negative control (not shown). Pictures are representative for 4 individual donors per group. Scale bars indicate 50 µm. (b) Western blot analysis from perilesional and lesional HS epidermis of three individual patients confirming stronger lesional p-JNK activation in lesional HS. LS, lesional skin; PLS, perilesional skin; PLE, perilesional epidermal lysates; LE, lesional epidermal lysates.
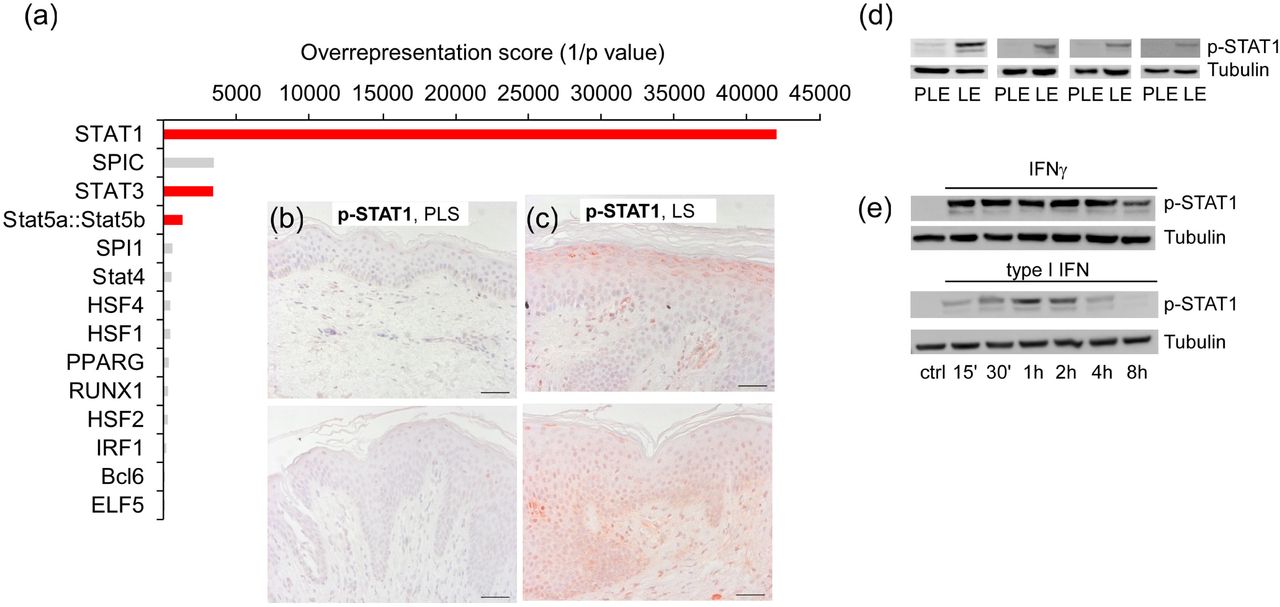
While a prominent JAK/STAT signature was already obvious from our large scale promoter analysis of all lesionally upregulated genes (Figure 3), a separate promoter investigation with smaller subsets of upregulated DEGs from individual overrepresented FACs uncovered an even more striking enrichment of STAT transcription factor sites. This was particularly true for STAT1 or STAT1:STAT2 sites that were highly enriched (p<0.001) in the promotors of the top 10 enriched FACs “Activation of immune response” (STAT1:STAT2; p ≤5.2E-5; STAT1; p ≤1.7E-4), “Neutrophil chemotaxis” (STAT1; p ≤8.0E-05), “Response to bionic stimulus” (STAT1; p ≤1.2E-4) “Cellular response to molecules of bacterial origin” (STAT1; p ≤1.0E-4), and “Response to cytokine” (STAT1; p ≤ 7.3E-4) (data not shown) but were also prominent in other top-enriched clusters such as “Innate immune response” (STAT1; p ≤2.4E-5; Figure 5, a). This apparent STAT1 signature in lesional HS epidermis could also be confirmed by immunohistochemistry (Figure 5, b-c) and immunoblot analysis (Figure 5, d). DEGs with well conserved IFNγ-responsive STAT-1 sites in their promoters also involved several of the lesionally most increased genes such as TNIP3, TLR8 and CXCL8 (Table S6), which consistently appeared in the top-regulated FACs (Table S3). STAT1 can be activated by several ligands including type I IFNs (IFNα and IFNβ) or IFNγ 33, which both have been implicated in the pathogenesis of HS 6,28. Western blot analysis of lysates from human epidermal keratinocytes (hEKs) isolated from lesional HS biopsies revealed a strong and sustained STAT1 phosphorylation upon IFNγ treatment and a transient activation with type I IFN (Figure 5, e). Thus, principally either IFNγ production or release of type I IFNs may explain the observed STAT1 activation in lesional HS epidermis.

(a) Overrepresented TF binding sites in the FAC “Innate immune response”. Red colored bars represent overrepresented STAT binding sites. Cut-off was set at an overrepresentation score >100.00 (p <0.01). (b, c) Anti-p-STAT1 immunohistochemistry of (b) perilesional or (c) lesional HS epidermis. Two representative images for stainings of n= 4 individual donors per group are shown; scale bars indicate 50 µm. (d, e) pSTAT1 immunoblots of total lysates from (d) perilesional and lesional HS epidermis of four individual patients, or (e) type I IFN- (10 U/ml) and IFNγ- (100 ng/ml) stimulated cultured hEK derived from lesional HS epidermis. hEK, human epidermal keratinocytes; PLS, perilesional skin; LS, lesional skin; PLE, perilesional epidermal lysates; LE, lesional epidermal lysates.
IFNγ–mediated STAT1 activation controls expression of immune-regulatory genes in lesional HS epidermis
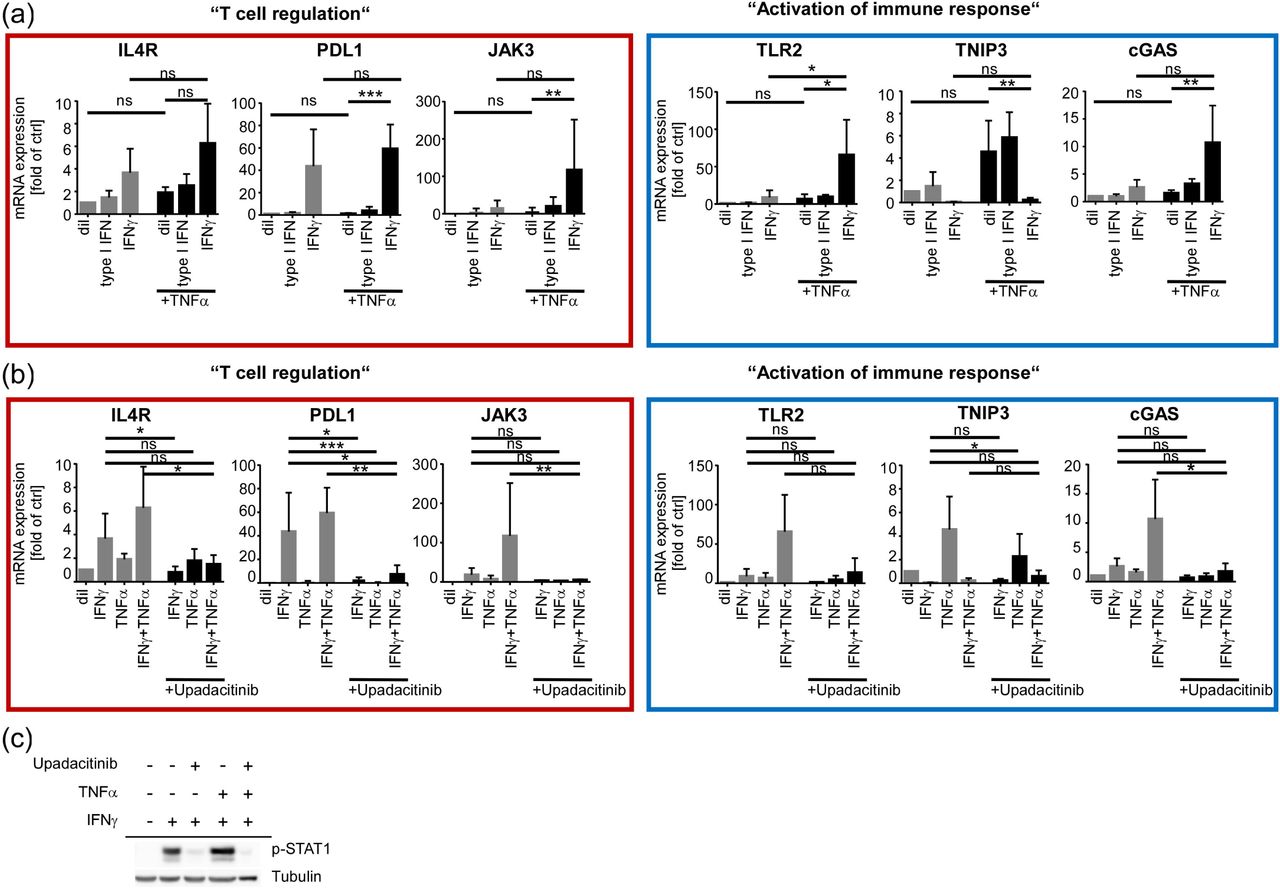
Our results raised the possibility that STAT1 activation may particularly influence induction of T cell-modulating genes or gene products controlling innate immune responses in HS lesions. To test whether stimulation with STAT1-activating IFNs may initiate expression of at least some of our previously confirmed lesional-specific DEGs, we isolated hEKs from lesional HS epidermis and stimulated them with either type I IFN or IFNγ. Additionally, we performed stimulations with TNFα, which has previously been implicated in the pathogenesis of HS 34 and included combinatory treatments of TNFα with either of the two IFNs to explore a potential cooperativity of both signals. Strikingly, the majority of our lesionally enriched test genes were transcriptionally induced by IFNγ but barely responsive to mono-treatment with TNFα or type I IFN. Only TNIP3 induction was clearly TNF-restricted (Figure 6, a). Notably, most test genes except for TNIP3, showed a trend towards increased mRNA abundance upon co-treatment with IFNγ and TNFα, suggesting that both cytokines may work in concert to induce a certain set of immune-modulating genes in HS epidermis (Figure 6, a). Treatment with the selective JAK1 inhibitor upadacitinib efficiently suppressed induction of all investigated genes but TNIP3 by IFNγ or IFNγ/TNFα cotreatment (Figure 6, b). Consistent with the prominent STAT1 signature characterizing several lesionally induced functional clusters of our RNA seq analysis, parallel Western blots confirmed efficient suppression of IFNγ- or IFNγ/TNFα-induced STAT1 phosphorylation by upadacitinib (Figure 6, c). This indicates that STAT1 activation critically contributed to the induction of our newly discovered HS-related keratinocytic IFNγ target genes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a, b) qPCR analysis of selected DEGs upon stimulation of HS-derived hEKs with (a) diluent (dil), IFN type I (10 U/ml), IFNγ (100 ng/ml), TNFα (50 ng/ml) or a combination of the respective IFNs with TNFα for 48 h, or (b) upon 48 h mono- or co-stimulation with IFNγ (100 ng/ml) and TNFα (50 ng/ml) +/- 250 nM of the JAK1-inhibitor upadacitinib. Data represent mean fold mRNA induction ± s.d. of the DEG in relation to GAPDH expression (at least n = 3 independent experiments with cells from different donors). *p≤0.05, **p≤0.01, ***p≤0.001; one-way ANOVA with post-hoc Dunnett correction. (c) Representative pSTAT1 Western blot with lysates of hEK stimulated in parallel.
DISCUSSION
Here we provide evidence that keratinocytes of lesional HS skin crucially orchestrate stress, innate and adaptive immune responses by inducing a complex AP-1- and STAT1-dominated gene expression pattern. DEGs with well conserved AP-1 or STAT1 sites in their promoters involved several of the top upregulated genes such DEFB4B, DEFB4A, TNIP3 and CXCL8, suggesting a prominent role of AP-1 and STAT1 signaling in epidermal HS responses. The observed AP-1 signature is likely a consequence of increased JNK activation in lesional keratinocytes, which may result from a general keratinocytic stress response due to excessive bacterial colonization. Accumulation of bacteria has previously been proposed to elict a strong stress signal affecting T cell responses 1 and innate immunity 35 in HS and may trigger activation of epidermal innate immune receptors such as TLR2 and/or production of TNF or IL-1β, which all are established triggers of JNK activation 36.
Unlike the rather unspecific AP-1 stress signature, the observed STAT1 phosphorylation probably indicates a specific response to high IFN levels in lesional HS epidermis. Both IFNγ released by effector T cells and type I IFN produced in response to neutrophil-induced damage have been implicated in the pathogenesis of HS 6,28 and could principally account for the observed STAT1 phosphorylation. Yet, two findings of our study argue against a major role of type I IFN in the epidermal response in HS. First, our large scale promoter analysis showed a strong overrepresentation of the more IFNγ-specific STAT1 sites in most regulated FACs but a less pronounced overrepresentation of STAT1::STAT2 sites that are more type I IFN-restricted 37-39. Secondly, we did not observe a sustained STAT1 phosphorylation and an appreciable induction of potential STAT1 genes by type I IFN stimulation of lesionally derived keratinocytes. In contrast, IFNγ stimulation induced both a sustained STAT1 phoshorylation and an upregulation of most of our test genes in vitro. Thus, overrepresentation of STAT1 sites in lesionally upregulated genes likely indicates a predominant response of lesional HS keratinocytes to IFNγ.
Strikingly, our RNA seq analysis and the subsequent bioinformatic evaluation revealed a clear overrepresentation of various T cell-relevant transcripts among the lesionally upregulated epidermal DEGs, which was confirmed by qRT-PCR analysis. While the keratinocytic function of many of those genes is unclear, we could show that most of them were strongly co-regulated by IFNγ and TNFα. Hence, their regulation in epidermal HS lesions above all may reflect a conserved epidermal responsiveness to high levels of IFNγ and TNFα contributing to T cell dysregulation, which has been proposed to initiate and sustain chronic inflammation in HS (Moran et al., 2017). On the other hand, the response pattern might also mirror the control of inflammatory processes in the dermis by lesional HS keratinocytes, which may inhibit T cell proliferation and cytokine production as recently suggested for allergic contact dermatitis 40. Particularly remarkable in this context is the observed strong induction of PDL1 in lesional HS epidermis. PDL1 is an established target of IFNγ-JAK1/2-STAT1/2/3-IRF1 signaling 41 and suppresses T cell immunity (proliferation, differentiation, cytokine secretion, cytolytic function) by binding to programmed cell death (PD)-1 42,43.
Besides the observed regulation of T cell-relevant genes, our RNA seq study also revealed a significant lesional overrepresentation of the functional gene cluster “activation of immune response” in HS epidermis. This cluster comprises several genes involved in innate immune activation that may instigate the apparently dominant T cell immune response in HS. Statistical upregulation of such genes might sensitize cells to endogenous and exogeneous danger signals thereby contributing to disease perpetuation and progression 44.
While NF-κB-dependent gene expression is critical for induction of an efficient immune response to bacterial infection 45, excessive or prolonged NF-κB signaling can foster inflammatory diseases 46. Therefore, the NF-κB pathway is tightly regulated by several intracellular proteins 47,48. These include TNIP3, an interaction partner of the ubiquitin-editing enzyme A20/ TNFAIP3, which counteracts inflammation by restricting NF-κB pathway signaling downstream of TNF and TLR4 49,50. NF-κB transcription factors were not among the top hits in the unfiltered promoter analysis of all regulated DEGs, albeit TNFα is associated with (auto-) inflammatory mechanisms in the pathogenesis of HS 51. The overexpression of TNIP3 might explain the low overrepresentation of NF-κB transcription factors in our upregulated gene set and contribute to chronification in HS by dampening NF-κB activity allowing innate immune-activating bacteria to persist in HS lesions to trigger a new cycle of recurrent inflammation.
The sensitivity of our verified IFNγ- and TNFα-inducible DEGs to JAK1/2 inhibition suggests a striking dependency of the epidermal gene response on JAK1/2 signaling. Considering that the currently only approved targeted therapy with the TNFα-antagonist adalimumab often results in unsatisfactory responses 52 these data raise the question whether JAK inhibitors might improve the success of non-surgical therapeutic approaches. Intriguingly, it was recently shown that anti-TNFα therapy resulted in a preferential attenuation of B cell-specific gene expression, whereas other parameters including IFNγ levels were unchanged after this treatment 53. Accordingly, suboptimal clinical response to anti-TNFα therapy in HS 52 might perhaps be due to a persistent IFNγ response making HS patients refractory to TNFα antagonists. At any rate, our results suggest that treatment regimens with JAK inhibitors - either alone or in combination with other agents – could be a promising new treatment option in HS.
Data Availability
Datasets related to this article can be found under EGA ID EGAS00001005544, hosted at EGA European Genome-Phenome Archive (https://ega-archive.org).
DATA AVAILABILITY STATEMENT
Datasets related to this article can be found under EGA ID EGAS00001005544, hosted at EGA European Genome-Phenome Archive (https://ega-archive.org).
AUTHOR CONTRIBUTION STATEMENT
Conceptualization: MS*, VGF; Formal analysis: LJ, VGF; Bioinformatical analysis: MS*, MS#; Funding Acquisition: VGF; Collection of clinical specimens: DP; Investigation: VGF, LJ; Methodology: MG, MS*; Project Administration: MS*; Resources: MG; Supervision: MS*; Visualization: VGF, LJ, MS*; Writing - Original Draft Preparation: MS*, VGF; Writing - Review and Editing: MS*, VGF, MG, LJ, DP, MS#.
*Marc Schmidt
#Mughda Srivastava
ACKNOWLEDGMENTS
We thank Andrea Knorz, Katharina Meder and Helga Sennefelder for excellent technical assistance. This work was supported by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF), University Hospital Würzburg, Germany (ZZ/25). We thank the Core Unit Systems Medicine at the Medical Faculty, University of Würzburg, that is co-funded by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) Würzburg for support of this work.
Footnotes
Funding sources: This work was supported by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF), University Hospital Würzburg, Germany (ZZ/25).
Conflict of interest: D.P. is member of the European Hidradenitis Suppurativa Foundation (EHSF) e. V. The Department of Dermatology, Venereology and Allergology, University Hospital Würzburg (with active members D. P. and M. G.) is a health care provider center of the European Network for Rare and Low Prevalence Complex Skin diseases (ERN Skin).
REFERENCES



