
Abstract
We present shaPRS, a novel method that leverages widespread pleiotropy between traits, or shared genetic effects across ancestries, to improve the accuracy of polygenic scores. The method uses genome-wide summary statistics from two diseases or ancestries to improve the genetic effect estimate and standard error at SNPs where there is homogeneity of effect between the two datasets. When there is significant evidence of heterogeneity, the genetic effect from the disease or population closest to the target population is maintained. We show via simulation and a series of real-world examples that shaPRS substantially enhances the accuracy of PRS for complex diseases and greatly improves PRS performance across ancestries. shaPRS is a PRS pre-processing method that is agnostic to the actual PRS generation method and, as a result, it can be integrated into existing PRS generation pipelines and continue to be applied as more performant PRS methods are developed over time.
Introduction
Genome-wide association studies (GWAS) provide a routine means of quantifying the effects of genetic variation on human diseases and traits. One possible use of these genetic effect estimates is the creation of polygenic risk scores (PRSs), an approximation of an individual’s genome-wide genetic propensity for a given trait or disease. Recent studies have shown that individuals in the upper extreme tail of polygenic risk for some common diseases have equivalent risk to those carrying monogenic mutations for these phenotypes1,2. Driven by these observations there is hope that polygenic scores can be used alongside traditional clinical and demographic predictors of disease to diagnose disease earlier and with greater accuracy3,4.
Unfortunately, the clinical utility of polygenic scores is currently limited by the GWAS on which they are based. The precision with which GWAS can estimate genetic effects on disease risk increases with sample size. Recent studies have suggested that most complex diseases will require somewhere between a few hundred thousand to several million cases to accurately capture genome-wide genetic effects on disease risk5,6. As a result, the information content of all current GWAS estimates is imperfect, reducing the accuracy of the polygenic scores generated from them. There is an expectation that GWAS meta-analyses across vast population biobanks will get us closer to quantifying SNP effects that fully capture heritability for some common complex diseases. However, many debilitating and life-threatening complex diseases have lower population prevalence, preventing even these large biobanks from ascertaining sufficient cases to facilitate the construction of accurate polygenic scores.
It is not only less common complex diseases that are set to be precluded from any clinical advantages brought about by polygenic scores. Genomics is failing on diversity7. On October 6th, 2021 the GWAS Diversity Monitor8 showed that 88.7% of individuals included in GWAS were from European ancestries. Recent studies have demonstrated the poor portability of polygenic risk scores across populations due to differences in effect sizes and LD structure9. Migration events and population bottlenecks can lead to large differences in allele frequencies between ancestries and, as a result of the biased application of GWAS, we are missing accurate disease risk estimates for the many variants that are only common outside of European ancestry groups10,11. Thankfully, the clarion call for major improvements in the ancestral diversity of GWAS, and genomics studies more generally, is now loud7,12,13. Recent studies in non-Europeans have highlighted the advantages of increased diversity of GWAS, delivering both novel genetic associations and biological insights that were missed even in the larger European GWAS studies9,14–16. If polygenic risk scores do start to deliver on their hype then further diversification cannot come soon enough – otherwise we run the risk of widening existing health inequalities.
While it is certainly true that genetic effects on disease can differ between populations, many risk variants are believed to be shared across divergent ancestry groups17,18. There is also a growing appreciation of the extent to which genetic effects are shared across different disorders. For clinically and biologically related diseases such as Crohn’s disease and ulcerative colitis, the two common forms of inflammatory bowel disease, genetic effects are often shared. Across immune-mediated disease more generally the number of known pleiotropic effects continues to grow, a phenomenon that is mirrored in other disease groups such as metabolic and psychiatric disorders. A principled pooling of information across traits19,20 and ancestries21–23 has already been shown to improve prediction accuracy of PRS. A common assumption of these methods is that weights given to each dataset are constant across SNPs. In reality, this assumption is frequently violated as the extent of sharing, either between two diseases or two populations, varies across SNPs24,25.
We introduce a novel method, shaPRS (pronounced Shapers), a PRS pre-processing step that can be integrated into existing PRS generation pipelines that allows integration of imperfectly shared information between two GWAS datasets. We assume one dataset is representative of the target population, hereafter referred to as the proximal dataset, and that a second adjunct dataset may provide relevant information but that the degree of relevance varies across the genome. Our approach, which only requires summary statistics for each dataset, estimates weights which summarise how relevant the adjunct dataset is at each SNP to perform a weighted meta-analysis of the two datasets. Where LD differs between the datasets, a blended pairwise SNP correlation matrix is used together with the weighted SNP effect estimates in any downstream PRS software. We show in large-scale simulations in the UK Biobank (UKBB)26 that shaPRS outperforms similar methods. We then apply shaPRS to six real GWAS datasets to illustrate the improvements it brings to PRS accuracy, both across diseases and across ancestral populations.
Results
Overview of method
shaPRS, which uses GWAS summary statistics, is a PRS pre-processing step based on a modified meta-analysis of two partially related GWAS studies. We begin by testing, at each SNP, evidence against homogeneity of effect between the two studies using Cochran’s test. From these test statistics, we calculate the local FDR (lFDR)27 as an estimate of the probability that the estimates reflect the same “common truth”. Where the lFDR is high, it is likely that the datasets can be combined and we favour β12, which is the standard inverse variance weighted average of the effect estimates in the proximal study, β1, and the adjunct study, β2. Our aim is to minimise variance by including information from the adjunct study, where doing so is unlikely to cause bias. Where the lFDR is low, we are conservative, and favour β1 from the proximal study, aiming to minimise bias at the expense of higher variance. We thus calculate a final shaPRS SNP effect estimate as
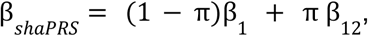 where π denotes the lFDR. As the use-case of our method is a seamless integration into existing PRS generation pipelines, a full set of summary statistics are derived, including standard errors, p-values and sample size, as described in the Online Methods.
where π denotes the lFDR. As the use-case of our method is a seamless integration into existing PRS generation pipelines, a full set of summary statistics are derived, including standard errors, p-values and sample size, as described in the Online Methods.
The current generation of most performant PRS generation methods28–30 also require an appropriate LD-reference panel. Therefore, to obtain an LD-reference panel appropriate for the derived summary statistics that represent information from different ancestries, we provide a method to derive a new matrix describing the correlation between βshaPRS across different SNPs (Supplementary Note).
Simulations of different trait, same-ancestry datasets
We performed simulations utilising common SNPs (MAF>1%) in the UK Biobank26 (UKBB) cohort. We compared shaPRS to two baselines approaches: single dataset analysis (β1 at all SNPs) and inverse variance weighted meta-analysis (β12 at all SNPs). The meta-analysis is equivalent to running shaPRS if there was no heterogeneity of effect anywhere across the genome, so allows us to examine the extent to which incorporating the measure of heterogeneity (lFDR) learned via the Cochran test improves PRSs. In recent years, several methods that exploit genetic correlation between related traits to improve association or prediction accuracies have been proposed including SMTPred20, MTAG19 and CTPR31. We choose SMTPred as a reference method to compare our novel approach against, as it also relies on only genome-wide summary statistics, thus it has an identical use-case to shaPRS. However, like other previously developed methods, SMTPred assumes a constant shared genetic aetiology across the genome. A detailed description of the simulation can be found in the Online Methods.
Genetic correlation (rG), which is a scalar metric, does not fully capture the overall structure of shared genetic aetiology. For example, a genetic correlation of 0.5 can be the result of all causal SNPs shared with a per-SNP effect correlation of 0.5, or alternatively, only half of the causal SNPs may be shared but with an effect correlation of 1.0. By fixing the genetic correlation at 0.5, but varying the fraction of shared and non-shared genetic effects we investigated and demonstrated the key ability of our method to adapt to such different compositions of overlapping genetic aetiologies. We also considered an additional scenario, where five SNPs contribute 5% of the total non-shared heritability for each trait. The rationale for including such SNPs was to model highly penetrant variants such as NOD2 in IBD25 or FLT3 in autoimmune thyroid disease24,32, which play an important role in differentiating these genetically overlapping traits from each other. In total, our simulations examined 108 different genetic architectures that arose from the examined parameters. The full set of parameters are summarised in Table 1, and Fig 1 presents a subset of our simulation results with an rG of 0.5 between the proximal and adjunct datasets. The full set of results from all scenarios can be found in Fig S2.

Heatmap of the squared correlation between simulated and predicted phenotypes for selected cross-trait genetic relationships. Warmer colours indicate better performance. a. Sample size N = 14,044, with a proximal/adjunct sample ratio of 50/50 or 20/80, a genetic correlation between proximal and adjunct traits of 0.5, no extra heterogeneity created by SNPs of large effect. p is the fraction of causal SNPs shared between the proximal and adjunct datasets, cor is the correlation of effect sizes between these SNPs. split is the ratio of the proximal to adjunct dataset sizes. b. The same scenario as a, with the addition of the extra heterogeneity created by five SNPs of large effect that contributed 5% non-shared heritability. Results across the complete set of simulated scenarios are shown in Fig S3.
The performance of shaPRS was better than any of the alternative methods in 94% of the simulated scenarios, frequently by large margins. ShaPRS’ capacity to accommodate genetic heterogeneity at a per-SNP level was demonstrated by a superior performance in scenarios where a given genetic correlation between two traits was concentrated amongst a subset of causal SNPs with stronger effect size correlations (See rG composition in Table1). As expected, shaPRS performed similarly to SMTPred20 in scenarios with a constant shared genetic aetiology (all causal SNPs shared between traits with weaker correlation in effect sizes) with no highly penetrant SNPs. The relative ordering of the performance of the methods did not change with the addition of the extra heterogeneity created by SNPs of large effect (Fig 1b and Fig S1b). However, such high penetrance variants further enhanced the advantage of shaPRS against all evaluated alternatives. In conclusion, our method compared favourably to both the baselines and SMTPred, which aims to exploit genetic correlation, particularly in scenarios when the underlying assumption of no non-shared SNPs with non-null effects was violated.
Application to inflammatory bowel disease subtypes
Inflammatory bowel disease (IBD) is a complex inflammatory disease of the gastrointestinal tract with a prevalence of 0.5% in Western countries33. Its two main clinical subtypes, Crohn’s disease (CD) and ulcerative colitis (UC) have a substantial but imperfect overlap in their genetic aetiologies, with a genome-wide genetic correlation of ∼0.5634. We performed a shaPRS analysis of ulcerative colitis (UC) and Crohn’s disease (CD) using an inflammatory bowel disease (IBD) GWAS dataset35 that included 3,765 and 3,810 UC and CD cases, respectively, and 9,492 shared controls. The Manhattan plot in Fig 2a illustrates how the estimated lFDR values capture the landscape of heterogeneity between UC and CD, with areas of highly incongruent effects (such as NOD2 on chromosome 16) featuring prominently among the peaks.

a. Manhattan plot depicting the genome-wide heterogeneity between Crohn’s disease and ulcerative colitis measured by Cochran’s Q test (Y-axis). Blue line represents SNPs with an lFDR < 0.5 and the red line represents SNPs with an lFDR < 0.01, which are also highlighted in green. b the performance of predicting the IBD subtype trained on the subtype alone, the combined IBD phenotype, shaPRS and SMTPred methods for Crohn’s disease (orange) and ulcerative colitis (green). Y-axis is the r2 between the predicted and observed phenotypes in a held out sample of sizes of 1,918/2,776 and 1,196/2,919 cases/controls, for CD and UC, respectively. The dots represent the 20 bootstrap samples built on the training set and evaluated on the held out test datasets, the bar is the mean across all bootstrap samples. The naming convention is as follows: ‘predicted:’ the target phenotype the PRS was evaluated on, and ‘trained:’ represents the method for training the PRS.
We built four sets of PRS. A set of baselines, trained either on cases consisting only of the single target subtype (CD or UC alone), or alternatively from the combined CD and UC cases (as an IBD phenotype), together with two advanced models, SMTPred and shaPRS. All PRS were built using LDpred2-auto29 as 20 bootstrap samples trained on our training set and evaluated on their respective test sets. We evaluated PRS performance on independent CD26,36 and UC37 cohorts, with 1,918/2,776 and 1,196/2,919 cases/controls, respectively (Fig 2).
We found that the performance, evaluated by squared correlation (r2) between predicted and observed phenotypes, of the PRS for predicting subtypes of IBD trained on the subtype itself versus the PRS trained on IBD were similar. From the point of view of the variance-bias trade-off latent in these experiments, these results make intuitive sense; we approximately doubled the sample size of the cases for traits that share approximately half their genetic aetiology (rG=0.56). Therefore, given this level of shared genetic aetiology, combining phenotypes to train PRS neither harmed nor improved the accuracy. However, we found that shaPRS substantially outperformed these baseline PRS. Evaluated against the proximal dataset alone, our method improved results by ∼23% and by ∼30%, for CD and UC, respectively. Compared to combining the CD and UC phenotypes, shaPRS increased performance by ∼14% and by ∼7%, for CD and UC, respectively. Additionally, shaPRS also outperformed SMTPred by ∼18% and by ∼17%, for CD and UC, respectively.
Leveraging datasets from different ancestries
GWAS have to date been concentrated in European populations, and the accuracy of PRS generated from one ancestry decreases in individuals of other ancestries, due to a combination of differences in LD, MAF, and causal variant effects between the training and test populations. We hypothesised that shaPRS could be useful to leverage information from GWAS in different ancestries. Therefore, to improve predictions in a proximal dataset, we leveraged information from adjunct datasets for the same trait in a different ancestry in a similar workflow as we did for different traits within the same population. Most state of the art PRS methods also require a relevant LD reference panel, therefore we derived one by blending the two original homogeneous SNP correlation matrices guided by the same blending factors as for the SNP effect estimates themselves (see Supplementary Note).
We evaluated our method by generating PRS using European ancestry summary statistics from the GWAS Catalog38 for five traits (asthma39, height40, BRCA41, coronary artery disease42 (CAD) and type 2 diabetes43 (T2D)), with adjunct association summary statistics from the BioBank Japan (BBJ) cohort44. These PRS were evaluated in a European ancestry subset of the UKBB cohort that did not overlap with any of the training data that the summary statistics relied on. Further details of individual studies and our data processing steps are described in the Online Methods.
We generated baseline PRS using the European GWAS only, and two PRS methods: PRS-CS and LDPred2-auto and PRS that leveraged information from BBJ using shaPRS combined with either PRS-CS or LDPred2-auto. We also evaluated our method against PRS-CSx23, a recently proposed method that integrates summary data from studies of populations of different ancestries that also takes into account MAF and LD differences. Unlike shaPRS, PRS-CSx is an all-in-one solution that performs both information pooling and the building of the final PRS profiles, but requires additional genotype level data from the target population to estimate hyperparameters. We provided these by using half the UKBB validation dataset to estimate the hyperparameters and the other half to validate all PRS. To ascertain how much of PRS-CSx’s performance is due to data from an additional genotype validation dataset, we also considered the performance of the European PRS from ‘stage 1’ of PRS-CSx (PRS-CSx-stage1), which relies only on summary information pooling without the weighting between the EUR and EAS PRS.
The performance of each PRS was evaluated by r2 and area under the curve (AUC) (for binary traits) between the predicted and observed phenotypes (Fig 3 and Table S1). Generally, shaPRS+LDpred2-auto, shaPRS+PRS-CS and PRS-CSx displayed a similar performance, with shaPRS+LDpred2-auto performing marginally better for three of the traits (T2D, asthma and BRCA). Each of these consistently outperformed the single dataset approach for every method and trait combination, except for PRS-CS and CAD, where PRS-CS alone performed similarly to the cross-ancestry methods. We also note that shaPRS consistently outperformed PRS-CSx-stage1, demonstrating its superior use-case in situations that have to rely solely on GWAS summary statistics.

Barplot of the results of the cross-ancestry analysis that compared the accuracy of six different methods to produce a PRS for EUR ancestry individuals. LDpred2 and PRS-CS are the LDpred2 method on auto option and the PRS-CS method, both trained on only the EUR datasets. shaPRS+LDpred2 and shaPRS+PRS-CS add preprocessing by shaPRS to leverage the EAS datasets whilst generating a EUR-specific PRS. PRS-CSx is the PRS generated by the PRS-CSx method that learns simultaneously from EUR and EAS datasets, and then uses additional genotype validation data from UKBB to create a weighted average of EUR and EAS PRS targeted to UKBB. PRS-CSx-stage1 is the EUR PRS generated by the PRS-CSx before the weighted averaging. This is included in the results to distinguish how much of the performance of the PRS-CSx method relies on information gained from joint learning from the summary data and how much is due to the weighted averaging with additional genotype data. a. Barplot of PRS performance evaluated by the area under the receiver operating characteristic curve (AUC) of the predicted and observed phenotypes. The error bars represent the 95% confidence intervals which were computed with 2,000 stratified bootstrap replicates. b. Barplot of PRS performance evaluated by the squared Pearson correlation coefficient (r2) between predicted and observed phenotypes. 95% confidence intervals were all too small to be visible at this scale. All PRS were evaluated on a strictly non-overlapping European ancestry subset of the UK Biobank.
Examining two of these examples in more detail helps to explain how shaPRS manages to increase accuracy compared to the single dataset analyses. ShaPRS adapts its behaviour to the pattern of genetic sharing in the studies (Fig 4). In either analysis, very few SNPs are detected to have genuinely different effects (i.e. low lFDR), but this proportion is greater amongst SNPs with significant effects and within the cross-trait compared to the cross-ancestry analysis. For the majority of SNPs with high homogeneity (lFDR > 0.5), standard errors are shrunk by shaPRS, whilst coefficients are also shrunk towards zero for non-significant SNPs (shaPRS p > 5×10−8) with higher homogeneity (lFDR > 0.5) but left unchanged otherwise. This is the same effect that would be expected for a meta-analysis. However, effect estimates change little at SNPs with high heterogeneity (low lFDR), which allows the specificity of individual dataset estimates to be leveraged when appropriate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Example of shaPRS analysis. The top row contrasts the distribution of effect heterogeneity measured by lFDR in a cross-ancestry analysis of asthma (left), and a cross-trait analysis of Crohn’s disease, leveraging a GWAS of UC as an adjunct dataset. a, b show the distribution of lFDR values, where low lFDR corresponds to higher heterogeneity in estimated effects. The bottom row compares the input beta (Beta_1) and standard error (SE_1) to its shaPRS-adjusted output (Beta_shaPRS, SE_shaPRS respectively) for the asthma analysis, divided SNPs according to whether SNP effect heterogeneity is low (c, d) or high (e, f). Colours indicate whether a SNP was detected to have a significantly non-zero effect (p < 5×10−8) in the shaPRS analysis.
Discussion
We have introduced shaPRS, a novel method that integrates genetic association information from heterogeneous sources and showed that it improves the accuracy of PRS for related traits and across ancestral populations.
A major strength of shaPRS is the ability to exploit the differential genetic architecture of related traits by considering the evidence for heterogeneity at each variant and weighting towards the estimate with the more beneficial properties: smaller variance in case of low heterogeneity or, alternatively, smaller bias in case of high heterogeneity. shaPRS can thus particularly improve the accuracy of a PRS when the genetic correlation structure between the proximal and adjunct datasets varies between SNPs. In our example of Crohn’s disease and ulcerative colitis, the pervasive sharing of genetic effects between the two diseases is well established45, and the genetic correlation between the two diseases has been estimated to be 0.5634. However, there are some SNPs with large differences in effect between Crohn’s and UC45; for example, in the NOD2 locus genetic variants explain around 1.5% of variance in liability of Crohn’s disease46, but there is no evidence of association to ulcerative colitis. More fully accounting for this inconsistent correlation in genetic effects between traits enables shaPRS to outperform competing cross-trait methods (as evidenced by a relative 14% improvement in the predictive accuracy of Crohn’s disease risk when leveraging data from UC using shaPRS, in comparison to training a PRS on the combined IBD phenotype). When applying our method to cross-ancestry prediction, shaPRS with either LDpred2 or PRS-CS performed at a comparable level to the cross-ancestry method PRS-CSx. A key advantage of shaPRS over PRS-CSx is that our method achieves a superior performance without the need for a validation genotype dataset matched to the target population (shaPRS always outperformed PRS-CSx-stage1). In practice we believe that this will often be the case for PRS aimed at individuals of non-European ancestries. Further, shaPRS is agnostic to the actual PRS generation method, thus it can be integrated into existing pipelines and continue to be applied as more performant PRS methods are derived in the future.
We structured our cross-ancestry examples to learn a European PRS, leveraging information from Japanese ancestry GWAS because this setup allowed us to evaluate performance in an independent (European) dataset. However, our expectation is that shaPRS will be more useful building PRS for non-European ancestries leveraging information from the generally larger GWAS from European ancestries, as suggested by simulations showing larger adjunct cohorts gave greater improvements in accuracy (Fig 1). In the coming years, to expand the clinical applicability of PRS, more ancestrally diverse populations will need to be recruited12,13. In the interim, methods such as the one presented here could contribute to more equitable health outcomes by leveraging existing datasets more efficiently.
Our simulations and real-world examples show that shaPRS can improve PRS estimation across a broad range of genetic architectures. While we have showcased the power of shaPRS for improving PRS estimates between traits and ancestries, this flexibility enables shaPRS to be applied whenever incomplete sharing of genetic effects is expected between two GWAS datasets. Other possible use cases for shaPRS could therefore include generating PRS for traits with heterogeneity of effect between the sexes or between different environments.
ShaPRS is designed to fit within existing pipelines as a pre-processing tool, thus, it is not in direct competition with other PRS generation tools such as LDpred229 or PRS-CS30. Our recommended approach is to pre-process GWAS summary statistics via shaPRS before taking them forward to a PRS tool of choice that would be used to produce the final profile scores. Finally, shaPRS also fits with the ongoing trend of reliance on summary statistics alone, without the need for access to genotype level data at any stage, as it provides a competitive performance without the need for a validation genotype cohort. Our method is open source and is freely available from https://github.com/mkelcb/shaprs.
Online Methods
ShaPRS genetic association summary statistics blending
Our approach is based on a weighted averaging of each SNP’s estimated effect between a single proximal dataset and an inverse variance meta-analysis of the proximal and adjunct datasets. The full derivations are set out in the Supplementary Note, and summarised here. Our method favours the proximal dataset effect estimate β1 where the effect estimates appear to differ between datasets, and combined effect estimate β12 (the standard fixed effects meta-analysis estimate obtained from β1 and the adjunct study coefficient β2) when the effect estimates for the two datasets are similar. In other words, we choose the more precise proximal phenotype (lower bias), where SNP effects are heterogeneous, but prefer the larger sample size (lower variance) where the SNP effects are congruent between single datasets.
To make this decision, we use Cochran’s Q-test to assess heterogeneity of effects between the two datasets at each variant, modified to allow for shared controls between the cohorts
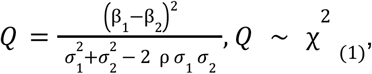 where σ1 / σ2 are the standard errors for the proximal and adjunct datasets, respectively and finally, ρ is an estimate of the correlation between β1 and β2 obtained as a simple function of sample sizes47.
where σ1 / σ2 are the standard errors for the proximal and adjunct datasets, respectively and finally, ρ is an estimate of the correlation between β1 and β2 obtained as a simple function of sample sizes47.
To estimate the probability that effects are heterogeneous, we used a local FDR approach, estimating
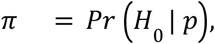 where H0 is the null hypothesis for the SNP, and p is the (adjusted) Q-test p-value obtained from the Chi-squared distribution with one degree of freedom as defined above. The lFDR values were then estimated from these p-values by the qvalue R package48.
where H0 is the null hypothesis for the SNP, and p is the (adjusted) Q-test p-value obtained from the Chi-squared distribution with one degree of freedom as defined above. The lFDR values were then estimated from these p-values by the qvalue R package48.
The blended effect estimate is then
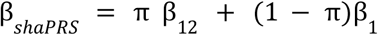
The goal of our method is to generate a new, complete set of summary statistics that may be used by a downstream PRS generation tool. These statistics include a new set of SNP coefficients, their standard errors and the correlation between coefficients. The Supplementary Note sets out derivations for the standard errors and correlation matrix, and functions to calculate these are provided in the R package https://github.com/mkelcb/shaprs.
Simulation analyses
Our simulations relied on the UKBB cohort, which has been previously described in detail elsewhere26. We excluded individuals who were sex-discordant, not ‘white British’ or had third-degree relatives in the cohort, as defined in the UK Biobank documentation. Genotype data were filtered to an intersection of the HapMap3 panel and a subset that excluded variants with an INFO score <0.8, MAF <0.1%, missing genotype rate >2% or a Hardy-Weinberg test P<10−7. From this subset, we randomly chose 31,598 individuals (twice the number of our IBD dataset).
The detailed simulation parameters were as follows. We evaluated the effect of cohort sizes by considering three scenarios, half, full and double the size of our IBD genotype datasets, which were 7,022, 14,044 and 28,088 individuals, respectively. 10% (3,510) of individuals were withheld as a test set that were not used for model training. We also considered two different ratios to split our source samples into the two phenotypes (proximal and adjunct). These ratios were 20/80 and 50/50 for phenotype 1 and 2, respectively. Additionally, we varied the range of pleiotropic architectures considered by evaluating three genetic correlations (0.1, 0.25 and 0.5) made up from three variations of shared and non-shared SNP effects. The motivation for the latter was to demonstrate the key ability of our method to adapt to different compositions of shared and non-shared genetic effects that comprise a fixed level of genetic correlation. We considered three different scenarios (low, medium and high, as defined in Table 1) of shared effects per genetic correlation, making up a total of nine arrangements. We also considered an additional scenario, where five SNPs contribute 5% of the total non-shared heritability for each trait. We used LDAK 5.049 to simulate 20 replicates for bivariate quantitative phenotypes whose SNP effect sizes we generated via our custom R scripts according to the schema described above for a total of 108 genetic architecture scenarios. We evaluated our method’s performance via comparing its predictive accuracy on the test set against three baselines, the single proximal dataset on its own, the meta-analysis of the proximal and adjunct datasets and the SMTPred method. SMTPred was trained directly on the PLINK summary statistics using its own ‘ldsc_wrapper’ function to estimate h2 and genetic correlations. To accommodate the scale of our simulations, the final PRS were generated via RapidoPGS, a light-weight PRS generation method50. To evaluate if using RapidoPGS had introduced any bias into our analyses, we re-generated the PRS of 40 randomly selected replicates (10 for each method) with LDpred2-auto. For this, we chose the scenario involving 14,044 individuals, phenotypes divided 50/50, with an rG of 0.5 made up from half of the causal variants shared with a correlation of 1.0, without any highly penetrant variants. We found that relative order of the performance of the methods did not change, and that the results were strongly congruent between LDpred2 and RapidoPGS (Spearman rank correlation of 0.795).
Inflammatory bowel disease dataset models
The availability of all IBD datasets are described under the Data and code availability section. The sample collection and initial quality control protocols are described in the original publications of each study35–37. The datasets were imputed via the internal Sanger imputation service utilising the merged UK10K + 1000 Genomes Phase 3 reference panel. The GWAS training datasets included 3,765 and 3,810 UC and CD cases, respectively, and 9,492 shared controls. The IBD dataset consisted of 7,575 UC and CD cases combined, and the same 9,492 controls. From this pool of data we derived 20 bootstrap samples using a combination of R and bash scripts. Starting from the HapMap3 panel, we filtered out variants based on the criteria of obtaining a Hardy-Weinberg equilibrium test p < 5×10−5 in controls or p < 5×10−7 in cases, INFO < 0.8, MAF < 0.1% or a missing genotype rate > 2%, which left 955,918 SNPs. Sex and 10 ancestry PCs were evaluated as possible covariates. The phenotypes were adjusted for covariates found to be significantly associated with the phenotypes in a multivariate logistic regression. Association statistics were obtained with PLINK via its ‘--assoc’ function. The PRSs for the IBD datasets were built using LDpred2-auto and the profile scores for our test set individuals were generated using PLINK’s ‘--score’ function.
Cross-ancestry datasets and PRS model evaluation
The Japanese association summary data for the five traits (asthma, height, BRCA, CAD and T2D) were all retrieved from the BBJ repository44,51. The European association data for the same five traits were sourced from different studies identified through the GWAS catalogue selected based on the criteria that they were of comparable sample size, and that they did not overlap with the (non-interim) UKBB release (Table 2).
To maximise the fraction of variants available across ancestries and summary datasets, HapMap3 SNPs were chosen that were shared between the Japanese and European summary statistics that were also present in the UKBB imputed dataset with an INFO score > 0.8. The final PRS were built after the removal of ambiguous alleles (A/T and G/C). PRS profiles were generated in PLINK52 and evaluated using individual genotypes from the UK Biobank cohort. For all traits we excluded related individuals and restricted the analysis to individuals with “white British” ethnicity (UKBB field 21000, code 1001). We also excluded ∼ 30,000 individuals which corresponded to the initial release and were genotyped with the BiLEVE array. We identified those individuals using field “22000” batches coded −1 to −11. For BRCA, CAD and T2D we applied the same selection criteria for cases and controls as previously described53, using the same UKBB codes for each of the relevant traits as in https://github.com/privefl/simus-PRS/tree/master/paper3-SCT/code_real). Briefly, we included as cases those individuals who self-reported the condition or were diagnosed by a medical doctor or the condition was included in their death record. For breast cancer we excluded individuals with other cancer diagnosis and restricted the analysis to females (108,21 cases, 147.134 controls). For T2D we excluded individuals with type 1 diabetes (12,288 cases, 301,822 controls) and for CAD we excluded individuals with other heart conditions (10,611 cases, 209,480 controls). For the asthma phenotype we identified individuals with the condition who had a positive response for self-reported code 20002_1111 (28,576 cases and 222,649 controls). For height we used 251,262 individuals in total with phenotype code 50.
After computing the PRS, for case control phenotypes we calculated the area under the curve (AUC) using the R package “pROC”, together with a squared correlation between the PRS and the measured trait (r2). Table 2 summarises the cross-ancestry PRS evaluation parameters.
Data Availability
ShaPRS R package is available from https://github.com/mkelcb/shaprs. Code to perform all analyses reported in this manuscript is available at https://github.com/mkelcb/shaprs-paper. The final PRS files and diagnostic data are available from the Supplementary data. The Crohn's disease and ulcerative colitis genotype data used here can be obtained via managed access athttps://ega-archive.org/studies/EGAS00001000924, https://ega-archive.org/studies/EGAS00000000084 and https://ega-archive.org/datasets/EGAD00000000005.
Declaration of interests
C.A.A. has received consultancy fees from Genomics plc and BridgeBio Inc. C.W. receives funding from GSK and MSD.
Funding information
This work was funded by the Wellcome Trust (203950/Z/16/A, WT220788, WT107881, 206194, 108413/A/15/D) and the MRC (MC_UU_00002/4) and supported by the NIHR Cambridge BRC (BRC-1215-20014). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
This research was conducted using the UK Biobank Resource under Application Number 30931.
Data and code availability
ShaPRS R package is available from https://github.com/mkelcb/shaprs. Code to perform all analyses reported in this manuscript is available at https://github.com/mkelcb/shaprs-paper. The final PRS files and diagnostic data are available from the Supplementary data. The Crohn’s disease and ulcerative colitis genotype data used here can be obtained via managed access at: https://ega-archive.org/studies/EGAS00001000924, https://ega-archive.org/studies/EGAS00000000084 and https://ega-archive.org/datasets/EGAD00000000005.
Acknowledgements
We thank Loukas Moutsianas for imputing the inflammatory bowel disease datasets using the Sanger imputation service. We thank all individuals who donated samples used in this study.
References



