
Abstract
We investigated autosomal mosaic chromosomal alterations (mCAs) in 10,248 non-small cell lung cancer (NSCLC) cases and 9,298 cancer-free controls of Chinese ancestry. Mosaic loss and copy-neutral loss of heterozygosity were associated with an increased risk of NSCLC, while mosaic gain was associated with a decreased risk of NSCLC, especially those spanning telomeres. The increased cell fraction of mCAs was also correlated with an increasing NSCLC risk in the affected individuals. Both multiplicative and additive interactions were observed between polygenic risk score (PRS) and the presence of mosaic loss, where carriers of mosaic loss events with cell fractions ≥5% among the high genetic risk group had the greatest risk for developing NSCLC. These findings suggest that mCA events may act as a new endogenous indicator for risk of NSCLC and have the potential to be jointly used with PRS to optimize risk stratification of NSCLC.
Introduction
Lung cancer is one of the most common incident cancers and the leading cause of cancer deaths worldwide1. The initiation and development of non-small cell lung cancer (NSCLC), the most common histology type of lung cancer, are believed to be influenced by a complex interplay between genetic architecture, aging, and environmental factors2,3,4. Genome-wide association studies (GWASs) have identified a number of inherited genetic loci for NSCLC5,6, and polygenic risk scores (PRS) constructed by combining these susceptibility loci have also been proved to be effective in quantifying individual risk of NSCLC6,7. However, the predictive performance of PRS differs among individuals depending on their age, lifestyles, or environmental exposures8,9,10, suggesting that the effect of heritable factors can be modified by non-heritable environmental risk factors.
In addition to tobacco smoking, a well-known risk factor for lung cancer11, recent evidence has linked several other environmental exposures to NSCLC risk, such as ambient air pollutants9,12, inhalable agents13, hazardous chemicals14,15,16,17 and unfavorable lifestyles11,18,19,20; however, the evaluation of associations between environmental factors and cancer risk is still challenging, as environmental exposures are temporally dynamic and difficult to measure accurately21,22,23, letting alone individuals who may have varied vulnerability and susceptibility in response to the same exposure24,25. Thus, an endogenous biomarker that could reflect internal effect of external environmental agents on biological capability is warranted to help yield efficient prediction of cancer risk.
Mosaic chromosomal alterations (mCAs) are large clonal structural somatic alterations detected in the whole blood-derived DNA, which are characterized by whole-chromosomal or sub-chromosomal level changes, including gain, loss, and copy-neutral loss of heterozygosity (CN-LOH). In addition to the crucial role of aging in the exponential increase of mCAs26,27, several other factors have also been reported to help shape the mCA landscapes, such as ambient air pollutants28, hazardous chemicals29,30, radiation31, and unhealthy lifestyles32,33,34,35, suggesting that mCAs can be viewed as an internal genomic signature associated with both internal and external exposures36. In the past decades, large population-based studies have established the effect of chromosomal mosaicism on early human embryogenesis37,38, birth defects39,40 and a broad range of adult diseases41,42 and cancers26,27,31,43,44,45. However, little is known about the type, frequency, and effect of acquired chromosomal anomalies to be associated with the development of NSCLC as well as their interplay with genetic factors.
Thus, in the current study, by using genotype array data of 10,248 NSCLC patients and 9,298 cancer-free controls of Chinese anscetry, we intend to systematically describe the landscape of mCAs in Chinese populations, investigate associations of detectable mCAs with risk of NSCLC, and examine their interplay with genetic factors (i.e., PRS).
Methods
Study populations
A total of 19,546 participants of Han Chinese ancestry were included in the current study, including 10,248 NSCLC cases and 9,298 cancer-free controls. The study population was derived from our recently published GWAS6 which included 4,149 cases and 3,198 controls from Nanjing and Shanghai, 2,155 cases and 2,035 controls from Beijing and Tianjin, and 3,944 cases and 4,065 controls from Guangdong. The demographics of these subjects were described in Supplementary Table 1. Of the included NSCLC cases, 6,839 were lung adenocarcinoma and 2,704 were lung squamous cell carcinomas. All lung cancer cases were histologically confirmed as new NSCLC cases by at least two local pathologists and were free of chemotherapy or radiotherapy before diagnosis. The included cancer-free controls were selected from a local community-based screening program for non-infectious diseases in Chinese populations. Written informed consent was obtained from all of the included subjects, and the study was approved by the Institutional Review Board (IRB) at Nanjing Medical University.
Genotyping and quality control
Blood-derived DNA of the included participants was genotyped with the Infinium Global Screening Array (GSA, version 1.0) that included 700,078 markers. Duplicate markers or SNPs were excluded from further analysis, if (1) with call rate <95%, (2) not in Hardy-Weinberg equilibrium (HWE, P<10−7 in controls or P<10−12 in NSCLC cases), (3) differed in frequency from the 1000 Genomes Project dataset, or (4) falling within segmental duplications with low divergence (<2%). Variant-level quality control was performed for each subpopulation independently. Finally, 581,940 (in Nanjing GSA GWAS), 581,252 (in Beijing GSA GWAS), and 605,292 (in Zhongshan GSA GWAS) variants on autosome chromosomes were included in the following analyses.
Haplotype phasing and detection of mCAs
The detection of mCAs was conducted on raw IDAT files from the Illumina GSA. Firstly, the genotype clustering was performed using the Illumina GenCall algorithm, and the resulting GTC files were converted to VCF files with the BCFtools gtc2vcf plugin (https://github.com/freeseek/gtc2vcf). The log2 R ratio (LRR) and B-allele frequency (BAF) values were used to estimate the total and relative allelic intensities, respectively. Then, SHAPEIT4 (version 4.2.1)46 was used for phasing across the three sample sets with default parameters, and the phased genotypes were ligated across overlapping windows using BCFtools concat plugin. Finally, the mCA calling was performed with Mosaic Chromosomal Alterations (MoChA)43,47 by leveraging long-range phase information to detect allelic imbalances across contiguous genomic segments. The identified mCAs were classified into copy number gain, loss, and CN-LOH based on the LRR and BAF values, and those that could not be assigned to any of the three types were designated as ‘undetermined’ (Supplementary Fig. 1).
Following previous studies, to filter out possible constitutional or inherited duplications, we excluded mCA events of length >0.5 Mb with relative coverage ≥2.5 or Bdev ≥0.1, and mCAs of length >5 Mb with Bdev ≥0.15. mCA events within the major histocompatibility complex (MHC) region (chr6:28,477,797–33,448,354, hg19) were further excluded due to the known propensity to call false-positive mosaic CN-LOH events within this locus.
The chromosomal location of mCA events was defined as follows: (1) events that only occurred around telomeric ends (±1 Mb from chromosome ends) were defined as telomere events, (2) events that crossed the centromere were defined as centromeric, (3) events that spanned an entire chromosome were defined as whole chromosome events, and (4) all other events were defined as interstitial.
Construction of polygenetic risk score
Based on all previously reported susceptibility SNPs (i.e., 81 SNPs in 40 loci) for lung cancer at a genome-wide significance level, our previous study constructed a PRS with 19 risk variants that are specific for Chinese populations6. Detailed information for the 19 SNPs included in the PRS was extracted from our previous study as described in Supplementary Table 2.
In the current study, the PRS was generated by summing all the 19 variants with the following formula: 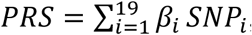 , where βi is the estimated weight (lnOR) of the ith SNP derived from our previous study6 and SNPi is the genotype dosage of each risk allele for each variant. The PRS was categorized as low (the first quartile), intermediate (25%∼75%) and high (the upper quartile) genetic risk (percentages were based on the distribution of the PRS among the cancer-free controls).
, where βi is the estimated weight (lnOR) of the ith SNP derived from our previous study6 and SNPi is the genotype dosage of each risk allele for each variant. The PRS was categorized as low (the first quartile), intermediate (25%∼75%) and high (the upper quartile) genetic risk (percentages were based on the distribution of the PRS among the cancer-free controls).
Statistical analysis
Logistic regression analysis was applied to evaluate the association between the presence of mCAs or specific mCA types and risk of NSCLC with adjustment for age, sex, smoking pack-years, DNA source, and ten principal components, in which odds ratios (ORs) and standard errors (SEs) were calculated. The multiplicative interaction was quantified by including a product term of genetic risk and the presence of mCA events in the model. The additive interaction was measured by calculating the relative excess risk due to interaction (RERI) and the attributable proportion because of the interaction (AP) based on coefficients of the product term. A Wilcoxon rank-sum test was used to assess the difference in the magnitude of different mCA types. All the reported P values were two-sided. All the analyses were performed using the R software (version 3.5.1, R Project for Statistical Computing).
Results
Characterization of the detected mCA events
Among the 10,248 NSCLC cases and 9,298 controls, we detected a total of 956 autosomal mCAs in 747 individuals (a prevalence of 3.82%), including 572 mCAs in 418 NSCLC cases (a prevalence of 4.08%) and 384 mCAs in 329 cancer-free controls (a prevalence of 3.54%) (Table 1). Of the mCA carriers, 657 had only one, 46 two and 44 at least three mCA events (Fig. 1a). Of the detected mCA events, the most frequent type was copy loss (36.40%, 348/956), whereas copy gain and CN-LOH constituted 23.01% (220/956) and 28.87% (276/956) of the mCA events, respectively (Table 1). In addition, most (54.71%, 523/956) of the mosaic chromosomal events began at a telomere and extended across some portion of the chromosomal arm, while only a small proportion (4.08%, 39/956) of mCA events spanned the entire chromosome, and 1.05% (10/956) crossed the centromere. The remaining mCA events were interstitial (40.17%, 384/956), spanning neither telomere nor centromere, and the chromosomal distribution of mCAs varied across different types (Supplementary Fig. 2).

(a) Distribution of the number of mCAs in each individual. Individuals with mosaic chromosomal gain, loss, or copy-neutral loss of heterozygosity (CN-LOH) are illustrated by different colors. (b) The genomic magnitude of mCAs by copy number alteration types. (c) Distribution of the number and proportion of mCAs with cell fraction ≥5%.
The size of detected mCA events was broadly consistent with those found in previous studies41,45, ranging from 16.97 kb to 249.25 Mb (median, 29.14 Mb) (Fig. 1b). The average magnitude of mosaic copy number gain events was significantly longer than that of mosaic CN-LOH (54.33 vs 31.57 Mb, P=1.25×10−8) and loss events (54.33 vs 21.99 Mb, P=8.38×10−13) (Fig. 1b). The estimated proportion of cells harboring an mCA event was widely distributed, ranging from 1.03% to 67.73% with a median of 7.53%, where a substantial number (N=623, 65.17%) of mCAs was seen with a cell fraction of ≥5%, particularly for mosaic loss events (N=276, 79.31%) (Fig. 1c). More than half of the mosaic gain (N=134, 60.91%) and CN-LOH (N=172, 62.32%) abnormalities also had a relatively larger cell fraction (Fig. 1c).
Demographic associations with mCAs
As age is a known risk factor for mCAs, we also observed an increased prevalence of autosomal mCAs with age in both NSCLC cases and cancer-free controls (Fig. 2, Supplementary Table 3). Interestingly, the proportion of mCA carriers was even higher among NSCLC cases than controls from age under 50 (2.61% vs 1.78%) to age greater than 80 (13.58% vs 7.97%) (Fig. 2), especially for the mosaic loss events (Fig. 2, Supplementary Fig. 3). No statistical association was observed for age at DNA collection with the magnitude or the clone size of mCA events (P>0.05) (Supplementary Fig. 4).

To evaluate the association of other common demographic factors with the incidence of mCAs, we compared the prevalence of mCAs among subgroups by sex and smoking status (Supplementary Table 3). Most mosaic gain events were enriched in males (1.23% vs 0.53%, P=4.73×10−7) and smokers (1.20% vs 0.78%, P=3.34×10−3), and mosaic CN-LOH events also tended to affect smokers (1.42% vs 1.14%, P=0.083), although not reaching a significance level. When age, sex, and smoking status were included in the same multivariable model, age still showed a significant association with the mCA frequency (OR=1.99, 95% CI=1.71-2.33; P=3.35×10−18), while males carried a significantly higher burden of mosaic gain events than females (OR=2.27, 95% CI=1.49-3.45; P=1.25×10−4) and smokers had significantly more mosaic CN-LOH events than life-long non-smokers (OR=1.46, 95% CI=1.04-2.05; P=0.031) (Supplementary Table 4).
Associations of autosomal mCAs with lung cancer risk
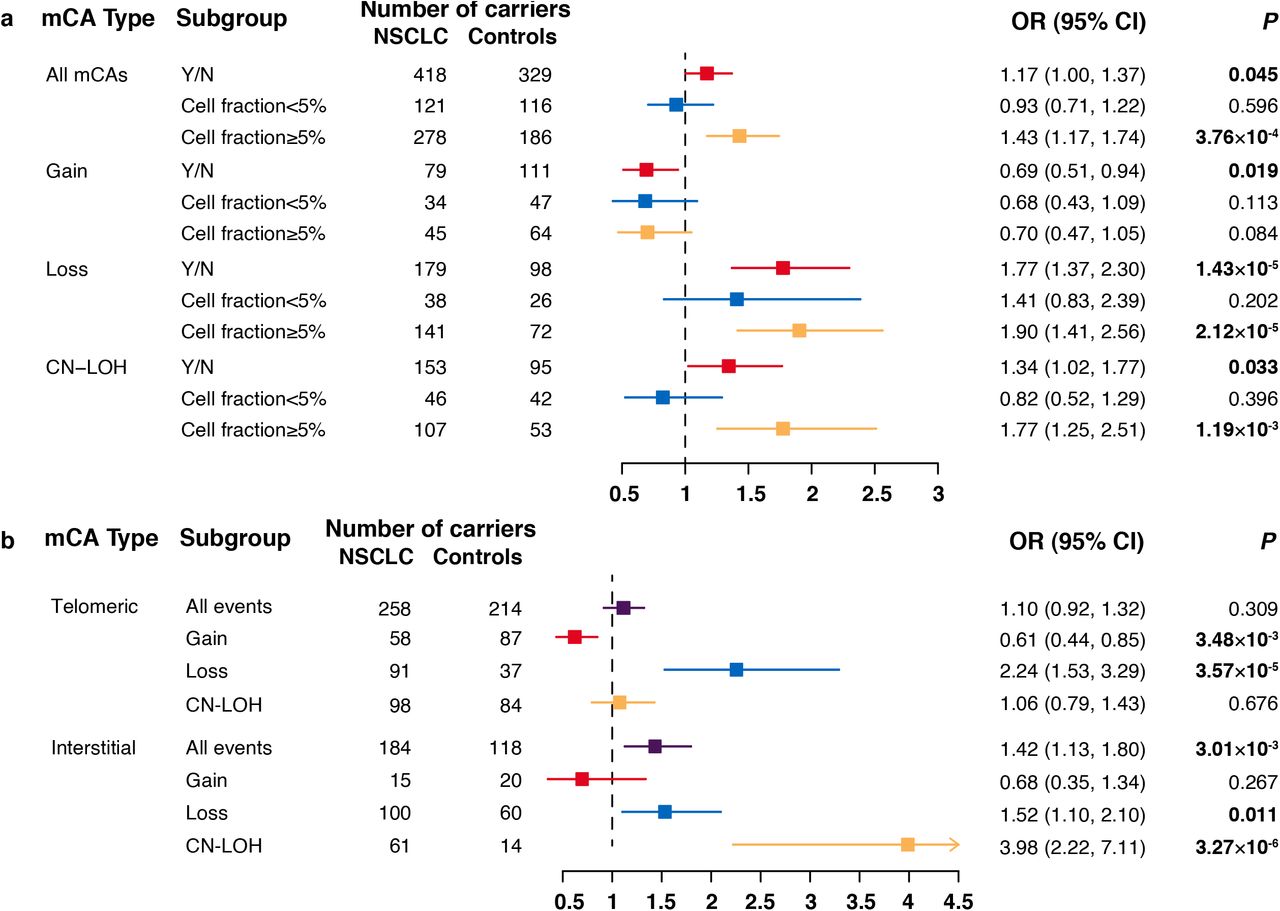
As the prevalence of mCA carriers among NSCLC cases was higher than that of controls across different age groups, we then evaluated the relationship between detectable autosomal mCAs and risk of NSCLC. We identified that the presence of mCA events was associated with a significantly elevated risk of NSCLC (OR=1.17, 95% CI=1.00-1.37; P=0.045) after adjustment for age, sex, smoking pack-years, DNA source, and ten principal components (Fig. 3a). Subgroup analysis by event type revealed that mosaic copy number loss had the strongest effect on NSCLC risk (OR=1.77, 95% CI=1.37-2.30; P=1.43×10−5), and mosaic CN-LOH also contributed to an elevated risk of NSCLC (OR=1.34, 95% CI=1.02-1.77; P=0.033) (Fig. 3a). In contrast, mosaic copy number gain was found to have a protective effect on NSCLC (OR=0.69, 95% CI=0.51-0.94; P=0.019) (Fig. 3a), and the effect was primarily observed in females (OR=0.34, 95% CI=0.16-0.72; P=4.87×10−3 for females and OR=0.84, 95% CI=0.60-1.19; P=0.333; PHet=0.032 for males) (Supplementary Table 5).

(a) Odds ratios (ORs) for the association between risk of NSCLC and mCAs by copy number alteration types. mCAs with cell fractions less than 5% or not are illustrated by different colors. Error bars indicate 95% confidence intervals (CIs). A total of 55 mCA events designated as ‘Undetermined’ failed in cell fraction estimation. (b) ORs for the association between risk of NSCLC and mCAs by genomic locations. Mosaic chromosomal gain, loss, or copy-neutral loss of heterozygosity (CN-LOH) are illustrated by different colors. Error bars indicate 95% CIs.
Then, we divided the mCAs according to their genomic locations (Fig. 3b, Supplementary Table 6) and observed that most (71.88%) of interstitial events were mosaic loss and CN-LOH, which all contributed to an elevated risk of NSCLC (mosaic loss: OR=1.52, 95% CI=1.10-2.10; P=0.011; mosaic CN-LOH: OR=3.98, 95% CI=2.22-7.11; P=3.27×10−6). Most of the mosaic gain events (77.00%) span across telomere regions, and had a protective effect on NSCLC (OR=0.61, 95% CI=0.44-0.85; P=3.48×10−3). As the effect of mosaic abnormalities on NSCLC risk may vary by cell fraction, we divided the mCAs according to their cell fractions, and identified that the presence of mCAs with large cell fractions (≥5%) had a much stronger association with the risk of NSCLC (OR=1.43 vs 0.93; PHet=0.012; Fig. 3a). In addition, we identified that the risk of NSCLC was much more strongly dependent on cell fractions (Supplementary Table 7).
Interaction between polygenic risk score and mCAs
Among the major interests in the current study is the joint effect of the presence of mCAs and genetic factors. To determine this, we first constructed the PRS of NSCLC with the 19 SNPs reported in our previous study (Supplementary Table 2). NSCLC cases tended to have a higher PRS (genetic risk) than those cancer-free controls (Supplementary Fig. 5). Interestingly, there was a synergetic effect of genetic factor (i.e., PRS) and mosaic loss events on risk of NSCLC in a dose-response manner (P for multiplicative interaction analysis =0.030) (Supplementary Table 8), where the overall risk of NSCLC increased as both PRS and the occurrence of mosaic loss events increased (Fig. 4). Specifically, compared to non-carriers of mosaic loss event with low genetic risk, carriers of mosaic loss abnormalities with a high genetic risk had the highest risk of developing NSCLC (OR=6.16, 95% CI=3.66-10.36; P=7.35×10−12) (Fig. 4), and the effect greatly increased in those who carry mosaic loss with cell fractions ≥5% (OR=7.64, 95% CI=4.01-14.56; P=6.25×10−10) (Fig. 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The associations between mosaic loss abnormalities and risk of NSCLC according to genetic categories (i.e., polygenic risk score). Abbreviations: OR, odds ratio; CI, confidence interval.
Significant additive interaction was also found between PRS and the presence of expanded mosaic loss (Supplementary Table 8): for carriers of mosaic loss events with cell fractions ≥5% among the highest genetic risk group, the RERI was 5.26 (95% CI=1.54-12.10), which suggests that there would be a 5.26-fold relative excess risk because of the additive interaction, accounting for 69% (31%-80%) of the risk in those participants who had both mosaic loss events and high genetic risk.
Discussion
In the current study, we comprehensively delineated clonal hematopoiesis in terms of copy number alterations among 19,546 individuals of Chinese ancestry and provided novel evidence for associations between mCAs and risk of NSCLC. We found that mosaic chromosomal abnormalities, especially mosaic loss of copy number, contributed to an elevated risk of NSCLC and that the effect increased as the cell fraction of the events increased. More importantly, we demonstrated a joint effect of mosaic copy number alterations and genetic risk, where the greatest relative increase of NSCLC risk was observed among those with expanded mosaic loss alterations and the highest level of genetic risk. Our findings provide novel evidence that mCA may serve as an endogenous genomic indicator for risk of lung cancer and have the potential to be jointly used with PRS to optimize the risk stratification for lung cancer.
Mosaic chromosomal alterations detected from blood-derived DNA are a group of somatic alterations that emerge in the period of early embryonic development and at the following stages of the lifespan48,49. Because this type of structural genomic alterations simultaneously spans dozens to hundreds or even thousands of genes, they have long been recognized for their necessity in hematological malignancy and mortality43,31,44. Recently, several studies proposed that mCAs could also be a possible source of unexplained susceptibility of solid cancers26,27,50; however, these studies have limited sample sizes not enough for cancer-specific analyses nor for copy number alteration type-specific (i.e., gain, loss, and CN-LOH) analyses. Our study demonstrated different effects of mCA types on NSCLC risk: while mosaic copy number loss and CN-LOH conferred an increased risk of NSCLC, the mosaic gain of copy numbers appeared to have a protective effect on NSCLC. Large copy number loss distributed across the genome was widely accepted as a risk factor of multiple cancers, no matter in the germline or somatic cells51,52,53. The mosaic gain, which primarily encompasses the telomere region, may help preserve genome stability and prevent the development of cancer.
Polygenic risk scores (PRSs) have advanced substantially in recent years and are widely used as an informative genetic measure for discriminating subpopulations at high risk of site-specific cancers6,7,54,55,56,57,58,59,60,61,62,63. Our recent work also constructed a PRS to stratify risk of lung cancer6. However, because many cancer types involve both environmental factors and genetic susceptibility, PRS in combination with other known risk factors will further improve risk prediction in the clinics. Here, we provided the first evidence, to the best of our knowledge, that mosaic loss of copy number synergistically with genetic factors increased risk of NSCLC and that approximately 69% of such an increased NSCLC risk could be attributed to their additive interaction, suggesting that incorporating mCAs into the existing PRS prediction model may contribute to a much improved predictive efficiency. In clinical practice, mCA may be expected as an ideal biomarker for risk assessment. Firstly, it could be a byproduct of the SNP array used for the PRS construction with no more tests43. Secondly, mCA can be monitored throughout life and provide a dynamic risk assessment coupled with PRS, given that mCAs provide a lifetime tracking of endogenous genomic alterations in response to environmental exposures26,64. With the development of electronic health records and the increasing availability of population biobanks, these findings should facilitate these resources to be effectively used to improve the precision risk prediction of lung cancer.
Although our results provide novel insights into associations of genomic mCAs with lung cancer, the interpretation of our findings needs to be considered within the following limitations. First, although this is the largest study for evaluating lung cancer risk associated with mCAs among Chinese populations, the number of detected mCA events is still not large enough to further investigate effect modification of genetic or environmental factors by such genomic alterations. In addition, the modest number of mCAs we obtained also precluded us from investigating properties of mCAs, such as different magnitude scales and chromosomal distribution with the incidence of lung cancer. Finally, although we observed a synergistic effect of genetic factors and mosaic copy number loss on risk of NSCLC, the case-control design used in the current study could not provide precise estimates of the risk. Further large-scale cohort studies are warranted to replicate our findings and refine the risk estimates.
In conclusion, the current study demonstrates that mosaic chromosomal alterations contribute to an increased risk of NSCLC, which greatly substantiates the effect of genetic factors on lung cancer and also provides novel insights into the precise prediction of lung cancer.
Data Availability
All the detected mosaic chromosomal alteration events and genotypic information for the 19 SNPs used for the calculation of polygenic risk score will be included as Supplemental Data before publication.
Author Contributions
ZH, CW, and NQ initiated and conceived the study. ZH, HS, GJ, HM, and JD assisted in data collection and recruitment of study participants. CC and LY assisted in sample collection and processing with SL, YX, YX, and JZ. NQ and CW developed the analysis plan and performed the bioinformatics/statistical analyses. NQ and CW wrote the first draft of the manuscript. All authors contributed to data interpretation and writing and approved the final manuscript for publication.
Declaration of interests
The authors have no conflict of interest to declare.
Acknowledgments
The authors would like to thank all the research staffs and students who participated in this work. This work was supported by the National Natural Science of China (81820108028, 81521004, 81922061, 82103926, 82072579), Research Unit of Prospective Cohort of Cardiovascular Diseases and Cancer, Chinese Academy of Medical Sciences (2019RU038), Natural Science Foundation of Jiangsu Province (BK20210534), and Postdoctoral Science Foundation of China (2021M691636).
References



