
Abstract
The APOE-ε4 allele is known to predispose to amyloid deposition and consequently is strongly associated with Alzheimer’s disease (AD) risk. There is debate as to whether the APOE gene accounts for all genetic variation of the APOE locus. Another question which remains is whether APOE-ε4 carriers have other genetic factors influencing the progression of amyloid positive individuals to AD. We conducted a genome-wide association study in a sample of 5,390 APOE-ε4 homozygous (ε4ε4) individuals (288 cases and 5,102 controls) aged 65 or over in the UK Biobank. We found no significant associations of SNPs in the APOE locus with AD in the sample of ε4ε4 individuals. However, we identified a novel genome-wide significant locus associated to AD, mapping to DAB1 (rs112437613, OR=2.28, CI=1.73-3.01, p=5.4×10−9). This identification of DAB1 led us to investigate other components of the DAB1-RELN pathway for association. Analysis of the DAB1-RELN pathway indicated that the pathway itself was associated with AD, therefore suggesting an epistatic interaction between the APOE locus and the DAB1-RELN pathway.
Introduction
Genome wide association studies (GWAS) have led to the identification of many genetic loci influencing the risk of dementia [1]. However, none of these approach the importance of the APOE locus [2] where the APOE-ε4 allele has a frequency of ∼15% in controls and has a risk ratio of >3 in cases. Other loci with allele frequencies of >1% have risk ratios of <1.4. Recent studies have shown that the APOE genotype is almost solely responsible for amyloid deposition whereas other components of Alzheimer’s disease (AD) genetic risk contribute to the occurrence of dementia in the context of amyloid deposition [3]. Furthermore, neuropathologic studies have shown that clinical diagnoses in Alzheimer series had a diagnostic accuracy of around 80%: this accuracy is implied by analyses comparing the large clinical GWAS with the smaller neuropathologic GWAS, leading to the concern that these larger GWAS are contaminated by other diagnoses. This concern is heightened by the reports of loci for frontotemporal dementia in case series labelled as Alzheimer’s disease in the most recent GWAS for the disorder [4].
With this background, we have undertaken an AD GWAS in individuals who are APOE-ε4 homozygotes for three reasons. First, because in this group diagnostic accuracy is very high; second, to assess whether in this context there is additional genetic risk at the APOE locus; and third, to assess which previously reported loci are replicated in these cases and whether there are any novel loci we can identify which are dependent on APOE-ε4 homozygosity. This study was possible in the UK Biobank [5] because it has a very large cohort, with a sufficient number (for statistical analyses) of APOE-ε4 homozygotes, where many participants are now reaching the age where they are at risk.
Here we report that the APOE allele alone accounts for the AD risk in the LD block on chromosome 19 in the European population. Furthermore, in APOE-ε4 homozygotes, we identify AD risk associated with the DAB1 gene that encodes a synapse regulatory protein. Subsequent analyses revealed a gene set association with the DAB-RELN pathway.
Methods
Phenotypes
Individuals from the UK Biobank were considered if they self-reported as white British and were of similar genetic ancestry by principal component analysis (UK Biobank field 22006), were unrelated (kinship coefficient < 0.04) and if they had not withdrawn consent to participate under UK Biobank. Participants were further excluded if they showed excessive missingness or sex chromosome aneuploidy, were outliers for heterozygosity, had mismatching self-reported and inferred sex from genotyping data, and had over 10 putative third-degree relatives. AD definition was derived using ICD-10 codes in hospital and death records. Individuals were coded as cases where dementia in Alzheimer’s disease (ICD-10 code F00) or Alzheimer’s disease (code G30) were present. Controls were defined as those without F00, G30, vascular dementia (F01), dementia in other diseases (F02) and unspecified dementia (F03). APOE status was assigned to each individual, as defined by SNPs rs7412 and rs429358 which are both present on the Affrymetrix Axiom genotyping array used. After quality control and restriction to APOE-ε4 homozygous individuals aged 65 or over, 288 cases and 5,102 controls were included in analysis.
Genetic quality control
The UK Biobank genetic data from the haplotype reference consortium (HRC), imputed by the UK Biobank [6], was restricted to biallelic SNPs (minor allele frequency > 0.05) with Hardy-Weinberg equilibrium > 10−6, INFO>0.4 and posterior probability>0.4. After quality control, 5,349,830 SNPs were included in analysis.
Analysis
Association analysis was conducted in PLINK2 [7] on UK Biobank dosage data using most recently recorded age, sex and the first 15 principal components (field 22009) as covariates. The significant findings (with the logistic regression) were further tested with Cox proportional-hazards regression (while controlling for the covariates) where the censoring occurred when a participant reported AD, allowing for the fact that some individuals have not reached the age at onset and may develop the disease given time.
The enrichment analysis of significant SNPs (at 5% significance level) or for SNPs showing the same direction of the effect (assuming that the chance to have the same direction of effect is 50%) was performed with binom.test() function in R.
The power calculations were performed with qnorm() function in R-statistical package at nominal 5% significance level (unless specified otherwise), where Z-score was estimated as log(OR)/var with the log(OR) as reported in the GWAS. In the Wightman et al. study [4], the largest OR was selected from the reported ORs in the list of contributing studies. The variance estimated as the inverse variance, with allele frequencies in cases and controls (corresponding to the SNP OR), and the sample size as in our study (N cases = 288, N controls = 5,102). Plots of regional associations were created using LocusZoom [8].
Epistasis was defined as deviation from joint two SNPs linear effects in the logistic regression model (known as statistical interaction). Significance of the interaction term was assessed using --epistasis option PLINK [7], accounting for the same covariates as above. The interaction plots were produced using matplotlib in python [9].
Gene-based analysis was run by MAGMA using FUMA v1.3.7 [10, 11]. Competitive setting of MAGMA was also used to test the candidate pathways for the enrichment of AD significant genes as compared to the rest of the genome.
DAB1-RELN pathway analysis
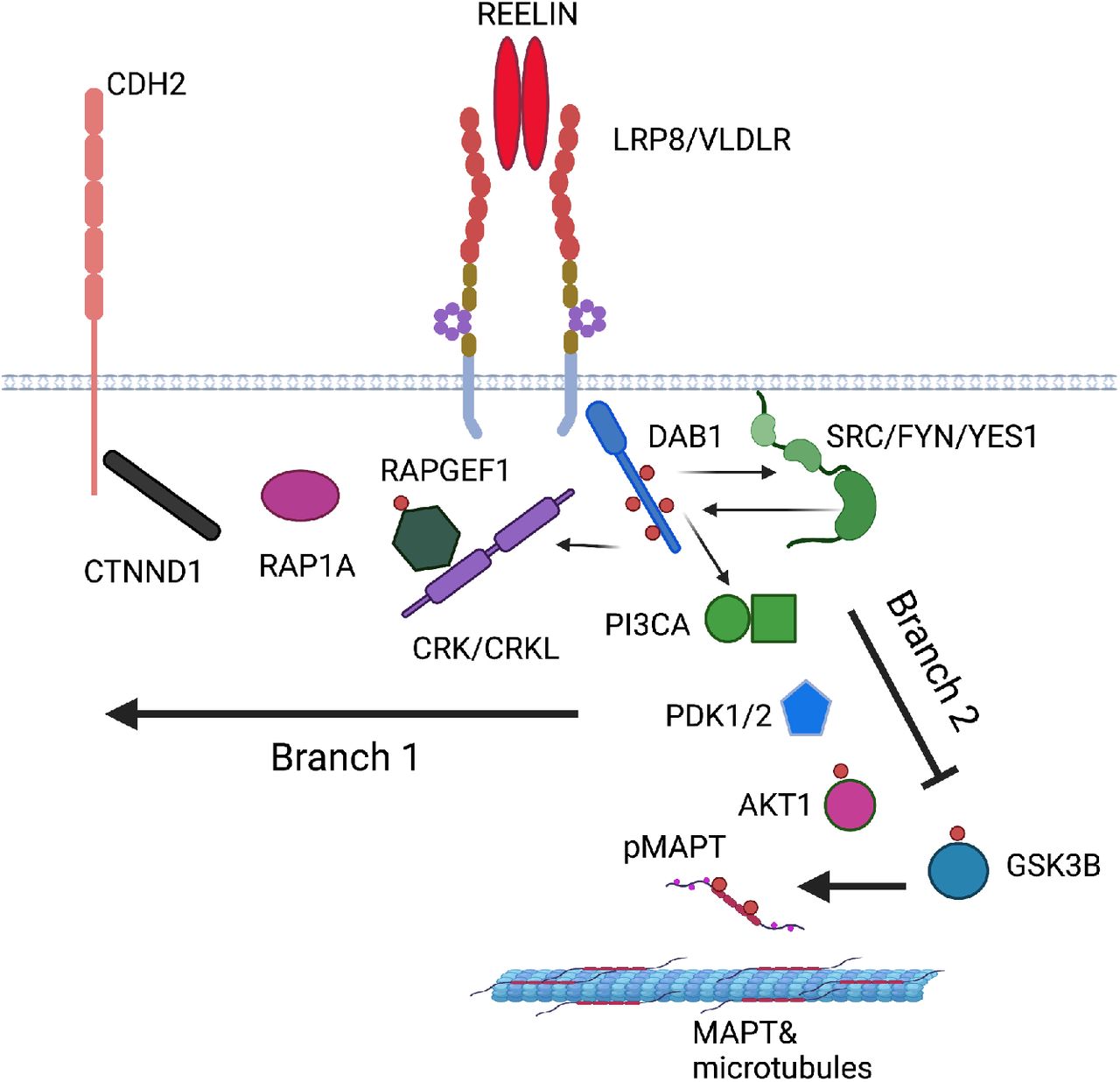
The canonical Reelin-Dab1 signalling pathway has been studied extensively in mouse neurons and brain [12]. For analysis, we divided the pathway into three sections: a) the receptor complex, (Reelin, the receptors ApoER2, VLDLR, the adaptor protein DAB1, and the tyrosine kinases SRC, FYN and YES) [13–16], b) branch 1 that regulates N-cadherin (CRK, CRKL, C3G, RAP1, P120 catenin, N-cadherin) [17–19] and c) branch 2 that is involved in microtubule-associated protein tau (MAPT) phosphorylation (PI3K, PDK, AKT, GSK3, STK25) [20–22]. We converted these mouse proteins to the homologous human genes with the BioConductor function in R and the NCBI database (www.ncbi.nlm.nih.gov/) yielding: a) RELN, LRP8, VLDLR, DAB1, SRC, FYN, YES1, b) CRK, CRKL, RAPGEF1, RAP1A, CTNND, CDH2, c) PIK3CA, PDK1, PDK2, GSK3B, AKT1, STK25.
Results
We present the results in the following order: (a) analysis of the APOE locus, (b) analysis of other previously reported GWAS in these cases, (c) identification of the DAB1 locus as a genome wide for disease, (d) assessment of other loci in the same DAB1-RELN pathway.
APOE locus
No suggestive variants were identified in the APOE gene or surrounding region (chromosome 19: 44.5-46.5 Mb, as defined previously [23]) with the lowest p-value at 0.003 within 1Mb of the APOE gene (Supplementary Figure S1) in APOE-ε4 homozygotes.
Other GWAS Hits
Loci previously reported as GWAS for association with Alzheimer’s disease status did not show a strong replication in the current analysis of APOE-ε4 homozygotes only (Supplemental Table S1). Though the power to detect the GWAS-reported effect sizes in this sample is not sufficient (see last column of Supplemental Table S1), four loci in CD33 (p=0.004), IQCK (p=0.009), LILRB2 (p=0.005) and SORL1 (p=0.007, MAF=0.04) had the strongest evidence for association in the current analysis and a consistent direction of effect between the current and previous GWAS. Weaker but nominally significant associations with the consistent direction of the effect were also observed in the APH1B (p = 0.024), BIN1 (p=0.011), SEC61G (p=0.015) and SNX1 (p=0.048) genes. In total, eight out of 77 SNPs (previously reported as genome-wide significant and available in our study), replicated at significance level with the same direction of association, which is statistically greater than chance (p=0.038). In addition, 53/77 (69%) SNPs have same direction of effect in the current analysis and previous GWAS which is greater than expected by chance (p = 0.001).
Identification of DAB1 as a locus
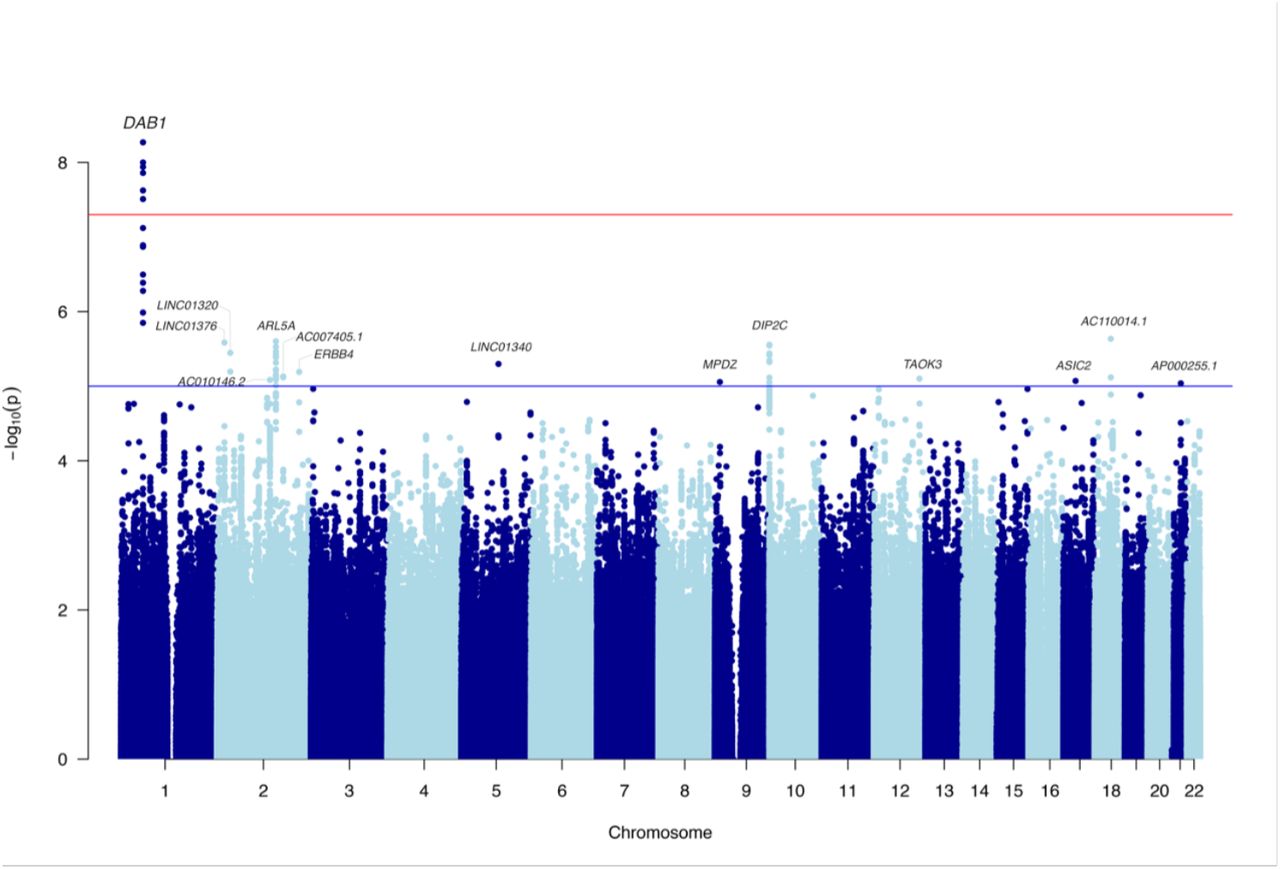
Multiple novel genome-wide significant intronic SNPs were present in DAB1 (lead SNP: rs112437613, OR=2.28, CI=1.73-3.01, p=5.36×10−9; Figure 1 and Supplemental Figure S2, Table 1). The minor allele T was associated with disease risk (MAF=6% in non-AD and 12% in ADε4ε4-participants of the UK Biobank). To allow for the fact that some individuals might not have reached the age at onset, we fit a survival regression model (adjusting for PCs and sex). The result remained highly significant (Hazard Ratio=2.27, CI=1.75-2.95, p= 7.8×10−10). The Kaplan-Meier graph (Figure 2) demonstrates that probability of getting the disease (y-axis) earlier (x-axis) is higher as the number of the risk alleles of rs112437613 SNP increases.
Novel genome-wide significant SNPs in DAB1.

Manhattan plot for the genome-wide association study in APOE-ε4 homozygotes with SNP MAF > 5%.

The cumulative risk of AD among APOE-ε4 homozygous of the UK Biobank participants, who carry 0, 1 or 2 risk alleles T the lead SNP (rs112437613) in DAB1.

Epistatic effect between APOE-ε4 and rs112437613 (DAB1) in the whole sample of the UK Biobank aged 65+ (N=229,748). All log(odds ratio) values are with respect to the baseline homozygote with no counted alleles at both loc. Orange/red bars have negative values. All odds ratios are adjusted for age, sex and principal components.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
REELIN-DAB1 signalling pathway based on studies in mouse neurons and brain (human protein names are shown, created with BioRender.com, see BioRender’s Academic License publication licence in the Supplemental material). The pathway branches downstream of the signalling complex. Branch 1 regulates the cell surface expression of CDH2 (N-cadherin) and branch 2 regulates MAPT phosphorylation.
The frequency of this allele is reported 4%-7% in European population cohorts (1000Genomes, TOPMED, GnomAD, Estonian, ALSPAC-UK, TWINSUK, Northern Sweden, see https://genome.ucsc.edu). However, this SNP (and others in LD with it) did not show even a nominal association to AD in recent GWAS that did not preselect for the ε4ε4 genotype: e.g. a study of 21,982 cases and 41,944 controls the p-values were p∼0.5 (see Table 1) [24]. Indeed, in a case/control sample (without screening for the APOE-ε4 status), the effect size of this SNP would be OR=1.016, as the proportion of cases, with both T allele of rs112437613 and ε4ε4, is 0.016 (=MAF(ε4)2*MAF(rs112437613 in ε4ε4) = 0.362*0.12, where 0.36 is the ε4 allele frequency in cases [25], and, similarly, of controls is 0.001. Therefore, the frequencies of the T allele in the overall sample are expected to be 0.061 in cases and 0.06 in controls, and consequently, the power to detect it with the sample size of the [24] study is close to 0 (∼3×10−7).
This observation led us to test for an epistatic effect in the whole sample of the UK Biobank aged 65+ (N=229,748). There was indeed significant epistasis between the two loci (p=1.5×10−5), whereas the effect of the T allele (rs112437613) was positive (OR=1.16, SE=0.11), but only nominally significant (p=0.021), providing evidence for cooperation between these two loci. The risk allele frequencies in this locus depending on APOE and AD status are shown in Table S2 and the risk of AD, depending on the genotypes at the two loci, is shown in Figure 5. The figure and table clearly show a statistical epistatic effect, where the disease risk is only visible in people with ε4ε4 genotypes.
Candidate analysis of other loci in the Reelin-DAB1 pathway
Dab1 encodes a cytoplasmic signalling adaptor that is predominantly expressed in neurons where it acts downstream of the extracellular ligand Reelin to regulate brain lamination during development [26–29]. Since Reelin-DAB1 signalling also performs an important role in the adult brain by promoting excitatory synapse maturation [30, 31] and modulating synaptic plasticity, learning and memory [32–35], we explicitly looked at the SNPs associations in the RELN gene (chr7:103,112,231-103,629,963). This gene is comprised of 2002 SNPs and the most significant SNP was rs171331137 (chr7:103479651) with OR=1.51 (SE=0.11), p=2.4×10−4 (Supplemental Figure S3). Similar to DAB1, we tested this SNP for interaction with APOE-ε4 in the whole UK Biobank sample. The interaction term was not significant (p=0.24), however the pattern of AD risk based on the pair of these markers was similar to DAB1 (Supplemental Figure S4).
The Reelin ligand and DAB1 adaptor proteins are bridged by two partially redundant transmembrane receptors APOER2 (LRP8) and VLDLR [15, 16]. Reelin binding to its receptors recruits DAB1 to their cytoplasmic tails activating the SRC family kinases, SRC, FYN and YES [13, 14, 36]. This leads to the increased tyrosine phosphorylation of DAB1 and the recruitment of additional signalling adaptor proteins that activate two key branches of the pathway (Figure 6). One branch is initiated by the binding of CRK and CRKL to phospho-DAB1, leading to the phosphorylation of C3G (RAPGEF1) and activation of RAP1 (RAP1A) [17, 18]. This leads to the upregulation of N-Cadherin (CDH2) cell-surface expression through engagement with p120 catenin (CTNND) [19]. A second branch is regulated by the binding of phosphatidylinostiol 3-kinase (PIK3KA) to DAB1 leading to the activation of PDK (PDK1, PKD2) and AKT (AKT1) ultimately suppressing the activity of the MAPT kinase GSK3 [20]. In mouse, deficiency of DAB1 has been shown to augment tau-phosphorylation and Stk25 has been implicated in this process [21, 22]. Since the signalling complex and the downstream pathways have potential significance in the development of AD, we tested their associated genes for enrichment in AD.
Using the results of the gene-based analyses described above, we tested whether the receptor complex and the two pathway branches contained significantly more AD associated genes as compared with the rest of the genome. We found that they were significantly (or almost significantly) enriched for genes associated to AD in the APOE-ε4 homozygotes (p-values 0.06, 0.009, 0.075, for the receptor complex and branches 1 and 2, respectively). The strongest significance was achieved when we combined the receptor complex and the two branches of the pathway (p=0.0055). Table 2 shows the details for each gene.
Results of the gene-based analyses for the genes in the DAB1-RELN pathway accounting for the number of SNPs and the LD structure for each gene using MAGMA software.
Discussion
No residual association at the APOE locus
APOE-ε4 is the strongest genetic risk factor for late onset AD. APOE-ε4 carriers have elevated risk for AD and earlier age-at-onset, with APOE-ε4 homozygotes at the highest risk [37, 38]. Many loci beyond APOE have been reported as associated with disease in increasingly large GWAS and meta-analyses, with over 80 susceptibility loci reported collectively [4, 39, 40]. We find no evidence to support the role of additional loci in an extended 2Mb region around APOE in APOE-ε4 homozygotes. This is supported by previous work on risk in the APOE region after adjusting for number of ε4 alleles [41, 42]. It is therefore unlikely that variants contribute additional risk to AD in the APOE region in APOE-ε4 homozygotes although association has previously been reported in PVRL2 and APOC1 in Chinese samples after adjusting for number of APOE-ε4 alleles [43]. Variants around APOE may explain additional variation in risk in populations where polymorphisms are in less pronounced LD with rs429358, and residual variability in APOE-ε3 carriers may still modify risk for the disease [44].
Other established GWAS hits
This study does not have statistical power to reliably determine whether all the previously reported GWAS hits are associated with disease in APOE-ε4 homozygotes or whether those which do show direct evidence for association (nominal significance) are grouped in any particular pathway.
Association with DAB1
Putative novel risk SNPs with strong evidence for association were mapped to the DAB1 gene on chromosome 1. Roles for DAB1 and RELN have previously been suggested in AD primarily based on studies in mice [36, 45–48] and functional genomic analysis in humans [49], but genome-wide association in humans has been lacking. However, it has been shown that the expression of DAB1 and RELN are altered in AD brains [50–52]. DAB1 interacts with Asp-Pro-any residue-Tyr (NPXY) motifs in the cytoplasmic domains of amyloid precursor protein (APP) as it does with similar motifs in the cytoplasmic tails of the Reelin receptors through its N-terminal PTB domain [53, 54]. The NPXY motif is required for APP internalization and its deletion reduces Aβ production [55]. DAB1 association with APP has been shown to reduce amyloidogenic processing [36], which suggests it is involved in the intracellular trafficking of APP. Reelin also reduces Aβ production in HEK293 cells that don’t express DAB1 [47]. In a mouse model of AD, heterozygosity of Reln increases the accumulation of Aβ plaques [45], suggesting that the pathway physiologically alters APP cleavage in a manner that would protect against AD. In addition, homozygous loss-of-function in Reln and Dab1 have been shown to augment tau-phosphorylation [21]. Reelin overexpression reduces abnormal somatodendritic localization of phosphor-Tau, Aβ plaques and synaptic loss in AD model mice [46, 48]. Thus there are links between the Reelin-DAB1 pathway and the two major pathological features of AD. In this study, both examined branches of the DAB1-RELN pathway had genes with significant association with AD. SNPs near RAP1A were significant; however, it remains to be determined if this branch regulates Aβ phosphor-Tau or another AD related pathology. The other major pathway downstream of Reelin-DAB1 has been associated with tau-phosphorylation and both AKT and PIK3KA from this branch were significantly associated with AD.
The dependence of the association between DAB1/RELN and AD on APOE-ε4 homozygosity is intriguing since there are several links between the Reelin pathway and APOE. The Reelin receptors are also APOE receptors and DAB1 binds the NPXY motifs in the cytoplasmic tails of other LDL-superfamily receptors [53, 54, 56], such as LDL-receptor related protein 1 that has roles in APOE/Aβ internalization and clearance [57]. Recent studies show that APOE-ε4 reduces recycling of ApoER2 back to the plasma membrane making the cells less responsive to Reelin [58] and that Reelin protects against the toxic effects of Aβ on synapses [59]. Thus in APOE-ε4 homozygotes, one can imagine a threshold effect with high APOE-ε4 driving a pathological cycle by reducing the effects of DAB1 and RELN signalling including its normal function to reduce Aβ production/toxicity and/or MAPT-phosphorylation.
While the effect the SNPs have on the function of DAB1 or other pathway genes remain to be determined, based on previous studies it would seem likely that they cause a partial loss-of-function that is potentially age dependent or cell-type specific in nature and would result in altered expression (eQTL) or splicing (sQTL). More than partial disruption of activity would likely lead to a developmental disorder in the homozygous individuals similar to loss-of-function alleles for Dab1 in mice and RELN in humans and mice [60]. The significant SNPs identified here fall in intron 2 and are found in 4-7% of the population. Interestingly DAB1 exomic variation is constrained and few variants are more prevalent than 1-2% (GnomAD) suggesting that the identified SNPs do not flag an alteration in the DAB1 coding sequence. DAB1 is alternatively spliced and differentially expressed most notably in a cell-type specific manner [26, 61–63]. Alternative splicing has been shown to regulate exons encoding a subset of the phosphorylation sites and a C-terminal exon altering Dab1 functionality in mice. We note that humans have a read through variant of exon 3 that would lead to transcriptional termination 14 residues later (variant 9) that has not been identified in mice. It encodes the first part of the phosphotyrosine binding (PTB) domain residues 37-69, but it is likely to be functionally inert since the PTB domain extends to residue 171 [64]. With this complexity and the size of the DAB1 gene, over 1 Mb, it could take significant effort to dissect the consequence of the SNPs identified here on gene function and AD.
In conclusion, we find a novel genome-wide significant hit in DAB1 in an APOE-ε4 homozygote AD GWAS. This seems to be a hit only in APOE-ε4 homozygotes. Furthermore, it seems that this association marks a more general importance of the DAB1-RELN pathway in disease pathogenesis. It is not clear why this pathway should be of such importance in APOE-ε4 homozygotes only, but a clue may be that such individuals have particularly dense Aβ pathology and one can imagine that this pathway either has a role in modulating APP processing or in driving tau-phosphorylation in a manner that is dependent on high Aβ levels. This work suggests that DAB1 has a protective role in late onset AD and highlights the importance of resolving the mechanism that likely involves the REELIN-DAB1 pathway for therapeutic development.
Data Availability
Data for this study was obtained under UK Biobank application 15175. Data is available from the UK Biobank at www.ukbiobank.ac.uk.
Funding
This work was largely funded by the UK DRI, which receives its funding from the DRI Ltd, funded by the UK Medical Research Council (UKDRI-3003), Alzheimer’s Society and ARUK. JH is supported by the Dolby Foundation, and by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. D.A.S. also received funding from the Alzheimer’s Research UK (ARUK) pump priming scheme via the UCL network. VEP is supported by Joint Programming for Neurodegeneration (JPND) - (MRC: MR/T04604X/1).
Competing interests
The authors report no competing interests.
References



