
Abstract
Rationale Chronic obstructive pulmonary disease (COPD) is characterized by pathologic changes in the airways, lung parenchyma, and persistent inflammation, but the links between lung structural changes and patterns of systemic inflammation have not been fully described.
Objectives To identify novel relationships between lung structural changes measured by chest computed tomography (CT) and systemic inflammation measured by blood RNA sequencing.
Methods CT scan images and blood RNA-seq gene expression from 1,223 subjects in the COPDGene study were jointly analyzed using deep learning to identify shared aspects of inflammation and lung structural changes that we refer to as Image-Expression Axes (IEAs). We related IEAs to COPD-related measurements and prospective health outcomes through regression and Cox proportional hazards models and tested them for biological pathway enrichment.
Measurements and Main Results We identified two distinct IEAs: IEAemph captures an emphysema-predominant process with a strong positive correlation to CT emphysema and a negative correlation to FEV1 and Body Mass Index (BMI); IEAairway captures an airway-predominant process with a positive correlation to BMI and airway wall thickness and a negative correlation to emphysema. Pathway enrichment analysis identified 29 and 13 pathways significantly associated with IEAemph and IEAairway, respectively (adjusted p<0.001).
Conclusions Integration of CT scans and gene expression data identified two IEAs that capture distinct inflammatory processes associated with emphysema and airway-predominant COPD.
Scientific Knowledge on the Subject Chronic obstructive pulmonary disease (COPD) is characterized by lung structural changes and has a prominent systemic inflammatory component, but the links between lung structural changes and patterns of systemic inflammation in COPD have not been fully described.
What This Study Adds to the Field We identified novel relationships between lung structural changes and systemic inflammation by simultaneously analyzing CT scans and blood RNA-sequencing gene expression using deep learning models. We identified two distinct Image-Expression Axes (IEAs) that characterize different inflammatory processes associated with emphysema and airway predominant COPD.
This article has an online data supplement, which is accessible from this issue’s table of content online at www.atsjournals.org.
Introduction
Chronic obstructive pulmonary disease (COPD) is one of the most prevalent chronic diseases (1), responsible for approximately 3 million deaths annually (2). COPD is characterized by persistent respiratory symptoms and poorly reversible airflow limitation (3). It is associated with an abnormal inflammatory response of the lungs to cigarette smoke or other noxious particles (4), which results in lung structural changes, including the loss or narrowing of airways (airway disease) and parenchymal destruction (emphysema) (5). In addition to its characteristic lung structural changes, COPD also has a prominent systemic inflammatory component (6,7).
Although lung structural changes and systemic inflammation are characteristic aspects of COPD, the origin of systemic inflammation and its relation to lung structural changes remain unclear (8). Computed Tomography (CT) scans provide a detailed characterization of lung structural changes (9), and blood gene expression captures systemic inflammatory signals (10). Therefore, we are motivated to understand the relationship between lung structural changes and systemic inflammation by applying a deep learning method to analyze CT imaging and RNA-seq data to identify novel relationships between lung structure changes and systemic inflammation patterns.
Based on the paradigm of COPD as a collection of treatable traits (11), we hypothesize that COPD heterogeneity can be described by continuous measures corresponding to distinct disease processes that are present to varying degrees in affected subjects. We refer to these continuous measures as “disease axes” (12), and we further hypothesize that integrative analysis of CT images and blood RNA-seq data can identify disease axes that identify patterns of association between lung structural abnormalities and systemic inflammation as quantified by blood RNA-seq. We tested these hypotheses by training a deep learning model on data from 1,223 subjects in the COPDGene study (13) with CT scans and blood gene expression data. Our analysis identified two disease axes that capture patterns of CT features consistent with emphysema and airway-predominant disease processes that are also associated with emphysema core-peel distribution and specific inflammatory pathways.
Methods
A comprehensive description of methods is included in the online supplement, and all analysis code is available in a GitHub repository (https://github.com/batmanlab/IEA).
Subject Enrollment and Data Collection
COPDGene enrolled 10,198 subjects with a minimum 10 pack-years lifetime smoking history at 21 centers across the United States (NCT00608764, www.copdgene.org) (13). Five-year follow-up data are available for 6,717 subjects, and 10-year follow-up visits are currently being completed. Subjects underwent spirometry, questionnaire assessments, standardized inspiratory and expiratory chest CT imaging, and genome-wide SNP genotyping. At the second visit (year 5), PAXgene blood RNA tubes were collected, and RNA-seq was performed. Each center obtained institutional review board approval, and all participants provided written informed consent.
Learning Image-Expression Axes (IEAs)
Only subjects whose CT scans were obtained on Siemens scanners with the b31f kernel were analyzed. CT features were extracted from DICOM image files using the following procedure. Each inspiratory chest CT scan was processed into 581 patches with a size of 323 mm3, following the practice in (14). For each patch, 128 features were generated using context-aware self-supervised representation learning (CSRL) (14), producing a matrix of dimension 581 × 128 for each CT scan.
Image-Expression Axes (IEAs) were constructed where CSRL features were the input to a multilayer perceptron (MLP) that produced a low-dimensional representation for each patch. Further supervised dimension reduction was performed to obtain subject-level Image-Expression Axes (IEAs) using a Product of Experts (PoE) model (15). At this stage, we applied statistically independent constraints with the Hilbert-Schmidt independence criterion (HSIC) (16) to ensure that each IEA captured an independent disease process. A final linear layer used IEAs as the input to predict the expression levels of the genes simultaneously. The parameters of the model were jointly optimized via Adam (17). In the training process, we evaluated the impact of feature selection on genes by testing each gene for the association with the top-128 principal components of the CSRL features in the training dataset. We also evaluated various thresholds for gene inclusion determined by the p-value of the F-test for each gene.
We randomly split the data into training and testing sets with sizes of 923 and 300, respectively. Model training was performed in the training set using five-fold cross-validation, giving us five models. The final IEAs were given by taking the average value of the IEAs from the five models.
Association of IEAs with Clinical Measurements
We computed Pearson correlation coefficients between IEAs and clinical measurements to understand their association. A full description of the measurements is included in the online supplement. Multivariable analyses were conducted by training Ordinary Least Squares (OLS) models for continuous measurements and logistic regression models for categorical measurements. We conducted a survival analysis (starting from the second visit in the supplement)with the Cox proportional hazards model(18). A full description of these models, including model covariates, is provided in the online supplement.
Comparison of IEAs to Other Disease Axes
We compared IEAs to the following disease axes: 1. COPD Factor Analysis Axes (FAs): Previously published phenotype disease axes identified through factor analysis(19). 2. PCA Image Only Axes (PCA-Is): Disease axes constructed by applying Principal Component Analysis (PCA) to the CSRL features. The comparative analyses included the calculation of Pearson correlation coefficients between IEAs and other disease axes, association analyses to clinical measurements, and comparison of nested models for clinical outcomes utilizing IEAs, FAs, and PCA-Is in determining whether IEAs improved the performance of models already containing FAs and/or PCA-Is.
Differential Expression and Usage Analyses
To identify the genes and pathways associated with IEAs, we performed differential gene expression analysis using limma and voom(20,21). Multiple comparisons were corrected with the Benjamini-Hochberg method to control the false discovery rate (FDR) at 10%(22). Gene Ontology (GO) pathway enrichment analysis was performed for pathways in the “Biological Process” category using the TopGO (v2.33.1) method (23). The threshold for statistical significance was an adjusted P-value < 0.001.
Results
One thousand two hundred and twenty-three subjects in the COPDGene study with complete CT scan and blood RNA-seq data were analyzed, and the flow diagram for the selection of subjects for analysis is shown in Supplemental Figure E1. The analyzed subjects were 50% female, 82% non-Hispanic white, and 18% African American, and the average age of the subjects was 67 years. The GOLD spirometric stage distribution of subjects was 42.5% GOLD 0, 44.0% in GOLD 1-4, and 13.5% with preserved ratio impaired spirometry (PRISm). For model training and validation, subjects were split into training and test sets, with no statistically significant differences in demographic or key clinical characteristics between these groups (Table 1).
Continuous variables are expressed as means and standard deviations. Categorical variables are expressed as percentages. P-values are obtained using the Kruskal-Wallis test for continuous variables and chi-square test for proportions, comparing the training and test data. FEV1 = Forced expiratory volume in 1 second; FVC = Forced vital capacity; perc15 = 15th Percentile Hounsfield unit in Inspiratory CT scan; %Gas Trapping: %LAA using −856 Hounsfield unit threshold on expiratory CT scan; Pi10 = the average wall thickness for a hypothetical airway of 10-mm lumen perimeter on CT; %WA segmental= the percentage of airway wall area for 3rd generation bronchi; GOLD: Global Initiative for Chronic Obstructive Lung Disease; PRISm=Preserved ratio impaired spirometry. , where the peel region is defined to be <5mm from the lung boundary and the core region is >20mm from the lung boundary. ΔFEV1 %predicted and ΔFEV1/FVC are computed by subtracting the visit 3 values from the visit 2 values of FEV1 % of predicted or FEV1/FVC and dividing them by the number of years between the two visits.
, where the peel region is defined to be <5mm from the lung boundary and the core region is >20mm from the lung boundary. ΔFEV1 %predicted and ΔFEV1/FVC are computed by subtracting the visit 3 values from the visit 2 values of FEV1 % of predicted or FEV1/FVC and dividing them by the number of years between the two visits.
IEA Model Training and Reproducibility Analysis
A schematic overview of the model training process is shown in Figure 1, and the data flow is summarized in Supplemental Figure E2. To maximize the stability of the IEAs and reduce the effects of sampling variability, we used nested cross-validation in the model training process to select the number of genes included in the model and the number of IEAs identified. For gene selection, we tested genes in the training data for the association to the top 128 principal components of the CSRL image features using an F-test, and a series of p-value thresholds for gene inclusion were explored, ranging from p = 1x10−6 to p = 1. The resulting IEAs were found to be stable across the entire range of p-value thresholds, and the threshold corresponding to p=0.01 was selected (Pearson’s r for IEAs across cross-validation folds ≥ 0.96, Supplemental Table E1). With this threshold, 4,685 genes were included in the final model. The number of IEAs identified by the model was determined by the amount of gene expression variance explained, which was the highest with two IEAs (Figure 2).

CT images were processed as 323 mm3 patches from which 128 features were constructed using the Context-Aware Self-supervised Representation learning algorithm (CSRL) (14). These features were the input for a multilayer perceptron (MLP) that processed a 581x128x923 tensor to output a 581x2x923 subject-level data representation. The subject-level latent representation (IEAs) is given by summarizing the patch-level features into a matrix of 2x923. We introduce a linear layer (2 -> 4,685) that estimates gene expression for each subject, taking the IEA as the input. We apply independent constraints to ensure IEAs are independent of each other. The overall objective function is given by minimizing the mean-squared error of the gene expression levels in prediction.

The figure on the left shows the plot of the 4,685 selected genes. The figure on the right shows the plot for all the genes. The figures show that when the number of IEAs is two, the total variance explained is maximized. We choose the number of IEAs to be two.
IEA Association to Clinical and Radiographic Features and Prospective Outcomes
To provide a clinical interpretation of the IEAs, we calculated their correlation to a range of COPD-related clinical and imaging measurements (Table 2). We refer to the first IEA as the emphysema axis (IEAemph), because it demonstrates a pattern of clinical associations consistent with quantitative emphysema. Specifically, higher levels of IEAemph were associated with lower lung function, emphysema, and lower BMI. The second IEA is consistent with airway disease and is referred to as IEAairway. Higher levels of IEAairway were associated with higher BMI, thicker airways, and less emphysema. The IEAs were uncorrelated with each other, suggesting that they may capture different underlying disease processes.
The symbols “*”, “**”, and “***” represent p<.05, p<.01, and p<.001, respectively. FEV1 = Forced expiratory volume in 1 second; FVC = Forced vital capacity; perc15 = 15th Percentile Hounsfield unit in Inspiratory CT scan; %Gas Trapping: %LAA using −856 Hounsfield unit threshold on expiratory CT scan; Pi10 = the average wall thickness for a hypothetical airway of 10-mm lumen perimeter on CT; %WA segmental= the percentage of airway wall area for 3rd generation bronchi; 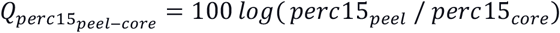 , where the peel region is defined to be <5mm from the lung boundary and the core region is >20mm from the lung boundary; ΔFEV1 %predicted and ΔFEV1/FVC are computed by subtracting the visit 3 values from the visit 2 values of FEV1 %predicted or FEV1/FVC and dividing it by the number of years between the two visits.
, where the peel region is defined to be <5mm from the lung boundary and the core region is >20mm from the lung boundary; ΔFEV1 %predicted and ΔFEV1/FVC are computed by subtracting the visit 3 values from the visit 2 values of FEV1 %predicted or FEV1/FVC and dividing it by the number of years between the two visits.
As shown in Table 2, both IEAs were negatively associated with emphysema peel/core distribution  , indicating that higher IEA values are associated with more emphysema in the core/central regions of the lung. To explore this association further, we performed sensitivity analyses, in which the lung was separated into concentric bands according to the distance to the lung boundary as described in the online supplement.
, indicating that higher IEA values are associated with more emphysema in the core/central regions of the lung. To explore this association further, we performed sensitivity analyses, in which the lung was separated into concentric bands according to the distance to the lung boundary as described in the online supplement.
To determine whether the IEAs provided clinical information in addition to standard demographic variables, we tested the significance of adding IEAs to regression models for various COPD-related measures (Table 3, Table 4, and Supplemental Tables E2 and E3). After adjusting for standard demographic variables, both IEAemph and IEAairway were significantly associated with FEV1 %predicted, FEV1/FVC, SGRQ total score, mMRC dyspnea score, and 6-minute-walk distance. IEAemph was also associated with Frequent Exacerbator (History), Frequent Exacerbator (Future), and overall mortality rate.
The table reports the β coefficients and corresponding 95% confidence intervals for IEAemph and IEAairway in linear models using the indicated COPD-related measurement or health outcomes as the response variable. All models were adjusted for age, gender, race, pack years, smoking status. The symbols “*”, “**”, and “***” represent p<.05, p<.01 and p<.001, respectively. FEV1 = Forced expiratory volume in 1 second; FVC = Forced vital capacity; perc15 = 15th Percentile Hounsfield unit in Inspiratory CT scan; %Gas Trapping: %LAA using −856 Hounsfield unit threshold on expiratory CT scan; Pi10 = the average wall thickness for a hypothetical airway of 10-mm lumen perimeter on CT; %WA segmental= the percentage of airway wall area for 3rd generation bronchi; 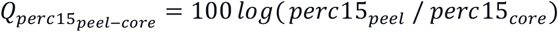 , where the peel region is defined to be <5mm from the lung boundary and the core region is >20mm from the lung boundary. ΔFEV1 %predicted and ΔFEV1/FVC are computed by subtracting the visit 3 value from the visit 2 value of FEV1 % of predicted or FEV1/FVC and dividing it by the number of years between the two visits.
, where the peel region is defined to be <5mm from the lung boundary and the core region is >20mm from the lung boundary. ΔFEV1 %predicted and ΔFEV1/FVC are computed by subtracting the visit 3 value from the visit 2 value of FEV1 % of predicted or FEV1/FVC and dividing it by the number of years between the two visits.
The table reports the β coefficients of the IEAemph and IEAairway and corresponding 95% confidence intervals from logistic regression models for frequent exacerbator status and a Cox proportional hazards model for mortality. All models adjusted for age, gender, race, pack years, smoking status as the covariates. The symbols “*”, “**”, and “***” represent p<.05, p<.01, and p<.001, respectively. Frequent exacerbator (history) indicated whether the subject had at least two self-reported exacerbations during the 12 months before the second visit. Frequent exacerbator (future) indicated whether the subject had at least two self-reported exacerbations during the past 12 months before the third visit.
To provide independent replication of our IEA associations, the IEA model was applied to 1,527 subjects from another subset of the COPDGene dataset that had not been used for model training. All the significant associations to clinical and longitudinal measures remained significant with very similar effect estimates indicating a high level of reproducibility for IEAs (Supplemental Tables E4, E5 and E6).
COPD Subgroups Defined by IEAs and Comparison to Existing COPD Subtypes
To further understand the clinical characteristics of COPD subgroups defined by IEAs, we divided the IEA space into four quadrants (Figure 3) and computed the average characteristics of each subgroup (Supplemental Tables E7). As expected, subjects with low IEAemph/low IEAairway values had the least obstruction (mean FEV1 89.2% predicted), the highest percentage of GOLD spirometric grade 0 subjects, low emphysema, and the thinnest airways. Subjects with high IEAemph/low IEAairway values had characteristics consistent with emphysema-predominant COPD, namely high emphysema and low BMI, with about 70% of GOLD grade 4 subjects present in this group. Subjects with low IEAemph/high IEAairway values had an airway-predominant profile with thick airway walls, elevated BMI, and the greatest proportion of PRISm subjects. Subjects with high IEAemph/high IEAairway had the highest SGRQ total score, highest mMRC dyspnea scores, and shortest 6-minute walk distance. In terms of COPD progression, the groups differed significantly in mortality risk (p<0.001) and frequent exacerbation status (2 or more exacerbations in one year) but did not change in FEV1. The group with the highest mortality was high IEAemph/high IEAairway followed by high IEAemph/low IEAairway. The latter group also had the highest percentage of subjects with frequent exacerbations, both for retrospective (p<0.001, chi-square test of all groups) and prospective exacerbations (p=0.048).

IEAemph is the emphysema axis, where higher values indicate more severe emphysema. IEAairway is the airway disease axis, where a higher value represents higher BMI and thicker airways. The space defined by these IEAs was used to stratify the cohort into four subgroups based on dividing the IEA space into four quadrants. Lung CT scans and clinical characteristics are shown for one subject in each quadrant, where the red mask represents the emphysema regions (< -950 HU). The characteristics of the four quadrants are summarized in Supplemental Figure E5.
To place IEAemph and IEAairway in the context with previously reported subtypes and disease axes in COPDGene, we compared these axes directly with the previously reported k-means subtypes (24) and factor-analysis derived disease axes (FAs) (19). In Figure 4, we observe that the highest values of IEAemph are found in the severe emphysema k-means subtype, and the highest values of IEAairway are found in the airway-predominant k-means subtype, confirming our clinical interpretation of these disease axes. Since the FAs also showed patterns consistent with emphysema (FAemph) and airway-predominant disease (FAairway), we compared the IEAs to the FAs and observed that IEAemph and FAemph showed a reasonably strong correlation (Pearson’s r = 0.58), but the IEAairway and FAairway axes showed only modest correlation (Pearson’s r = 0.28, see Supplemental Table E8). Examination of the pattern of clinical associations for IEAairway and FAairway revealed that while airway axes were positively correlated to airway wall thickness, IEAairway is negatively correlated to emphysema whereas FAairway is positively correlated (Supplemental Table E9). To determine whether the IEAs provided additional information about COPD phenotypes (FEV1 % of predicted, FEV1/FVC, SGRQ, mMRC, retrospective frequent exacerbations, and 6-minute walk distance) and COPD progression (mortality and prospective frequent exacerbations) above and beyond FAs, we constructed baseline models for each COPD phenotype and progression measurements with FAs included and then compared them to models including both FAs and IEAs. In most cases the models with IEAs included outperformed the baseline models (p<0.001 for all COPD phenotypes and mortality, Supplemental Tables E10 and E11).

P-values are obtained using the Kruskal-Wallis test. The symbols “*”, “**”, and “***” represent p<.05, p<.01, and p<.001, respectively, for a t-test comparing to the Relatively Resistant Smokers Group. No symbol indicates non-significant results.
Comparison to PCs based on Images Alone
After observing that IEAs contain additional clinically relevant information relative to standard features extracted from CT images, we sought to determine whether the additional information came only from applying dimension reduction to the CT images (CSRL features), or whether there was added value from our algorithm that combined the CT features with gene expression. To make this comparison, we constructed disease axes from images only by using PCA to extract the top-2 PCs of the CSRL features, denoted as PCA-Is. We then compared the predictive performance of linear models that utilize both IEAs and PCA-Is with the nested version that involves the PCA-Is only, and we observed that the models including IEAs were superior to models with PCA-Is only for all of the six studied COPD phenotypes as well as prospective exacerbations and mortality (p<0.001, Supplemental Tables E12 and E13). These results suggest that by incorporating gene expression data during training, IEAs extract more clinically important information than similar methods that utilize imaging features only.
IEAs are associated with inflammatory pathways
To understand the biological aspect of the IEAs, we first confirmed that IEAs explained a greater proportion of gene expression variance that PCA-Is, as demonstrated in Figure 5, which shows that IEAs explain a greater proportion of variation on a per-gene basis than PCA-Is (557 genes with R2 >10% for IEAs versus 68 genes with R2 >10% for PCA-Is).

The figure on the left shows the histogram for the 4,685 selected genes. Within these genes, there are 557 genes with R2 >10% for IEAs and 68 genes with R2 >10% for PCA-Is. The figure on the right shows the histogram for all the genes (18,487 genes in total). There are 622 genes with R2 >10% for IEAs and 69 genes with R2 >10% for PCA-Is.
To identify specific biological processes associated with each IEA, we performed differential expression and pathway enrichment analysis. We identified 6,494 and 3,815 genes associated at an FDR of 10% with IEAemph and IEAairway, respectively (Supplemental Tables E14 and E15). Gene Ontology pathway enrichment identified 29 and 13 enriched pathways (p-value < 0.001) for IEAemph and IEAairway, respectively (Supplemental Tables E16 and E17). The most significantly associated pathway for IEAemph was neutrophil degranulation, whereas IEAairway had the strongest enrichment for RNA processing. The most significant pathway results are shown in Table 5.
Gene ontology (GO) pathway enrichment analysis was performed using the GO “Biological Process” gene sets with p-values calculated with the Fisher exact test statistic using the weight01 algorithm in topGO(23) (v2.33.1) that accounts for dependency in GO topology.
Discussion
In this paper, we used deep learning to identify novel connections between lung imaging features and blood gene expression. The deep learning model provided novel disease axes, i.e. IEAs, that captured elements of shared variability between CT scans and blood RNA-seq, and we demonstrated 1) that these IEAs are associated with important COPD-related physiologic and functional measures, 2) that these associations contained information that is independent from pre-existing, standard clinical and imaging variables, 3) that the IEAemph axis is significantly associated with prospective mortality in multivariable models, and 4) that IEAs capture distinct patterns of connection between lung structural changes and systemic inflammation.
Many of our main results are consistent with our current understanding of COPD. IEAs capture the two cardinal pathologies of COPD, emphysema, and airway disease; but clearer links between these aspects of lung structure and systemic inflammation emerge from the joint analysis of CT images and blood RNA-seq. First, neutrophilic inflammation was strongly associated with emphysema but not the airway axis. This agrees with the prominent role of neutrophils in alpha-1 antitrypsin associated (AAT) emphysema(25), and it provides further support for the role of neutrophils in the emphysema of “typical” COPD. There is evidence as well for a role for specific adaptive immune processes that show significant enrichment for both IEAemph and IEAairway, though the inflammatory signals that we observed in blood differ from the B-cell predominated signatures that have been observed in some lung transcriptomic studies of emphysema(26). The biological pathway enrichments we observed are consistent with previous reports of the association of emphysema to biomarkers related to systemic inflammation, oxidative stress, and elevated plasma fibrinogen levels(27). IEAairway is strongly correlated to BMI, which coincides with a previous hypothesis that obesity-related adipose tissue hypoxia and systemic hypoxia due to reduced pulmonary function contribute to the systemic inflammation of COPD(28). Future studies, including single cell transcriptomic data, could better identify the association of emphysema and airway disease with specific types of innate and adaptive inflammatory cells.
While our IEAs seem most descriptive of emphysema and airway disease, they are not completely correlated to standard CT measurements of emphysema and airway disease, and they differ notably from machine-learning disease axes based on imaging alone (PCA-Is) or based on imaging and spirometry (FAs) (19). While none of these representations of emphysema and airway disease is demonstrably superior to the others, the IEA axes have a clear interpretation due to the integrative nature of the deep learning algorithm, whose goal was to find shared variability between CT images and transcriptomic patterns in the blood. The clinical relevance of these axes was demonstrated through regression models showing that IEAs were significantly associated with a wide range of COPD-related measures, including mortality. In the future, these algorithms can be extended to incorporate additional sources of molecular or imaging data.
While the IEAs primarily captured patterns of emphysema and airway-predominant COPD, they were also significantly correlated with core versus the periphery (“peel”) emphysema distribution. Previous work has demonstrated numerous clinically relevant associations to aspects of emphysema distribution, most notably for core/peel and apical/basal emphysema distribution(29-33), and a machine learning analysis of images alone also identified peel-core emphysema distribution as an important dimension of COPD-related variability(34). Our analysis suggests that systemic inflammation is most strongly associated with core/peel rather than apical/basal emphysema distribution, and that the amount of emphysema in the core region is associated with the more severe disease along both the IEAemph and IEAairway axes. Since quantification of the lung peel can be influenced by technical factors related to lung segmentation, we conducted sensitivity analyses that confirmed a consistent association for the IEAemph axis, whereas the IEAairway association was clearly present only when the analysis included the outermost lung regions. Accordingly, we have high confidence in the IEAemph association to core/peel distribution, but it is not clear whether the IEAairway axis association reflects a true biological relationship or technical artifacts.
By collecting CT scans, blood transcriptomics, and detailed phenotype data on thousands of current and former smokers enriched for COPD, the COPDGene Study provides a novel opportunity for the application of machine learning to better understand the connections between the lung structure in COPD and molecular mechanisms of systemic inflammation. Like all machine learning models, the construction of our model required many explicit and implicit design choices. In our model, we extracted patch-level representations via self-supervised learning. Such methods are capable of extracting generalized and semantically meaningful features(35). The linear independence assumption in our model was intended to identify IEAs that captured distinct underlying disease processes and increase the reproducibility of our model. Unlike previous studies that explore the relationship between COPD imaging and omics by associating previously discovered image patterns to omics data(24,36,37), our method potentially identifies new image patterns that have not been previously explored.
The main strengths of this study are: 1) The joint analysis of full DICOM data from CT images and gene expression data via deep learning is novel and provides new biological and clinical insight into COPD. 2) The sample size is large, allowing for more power to identify novel discoveries. 3) We used a number of techniques to improve the reproducibility of our disease axes, including cross-validation, sensitivity analysis, and the use of constraints in our modeling procedure. The main limitations are: 1) Our study is limited to blood RNA biomarkers, which capture systemic inflammation but no other important aspects of the COPD inflammatory response, such as lung gene expression and protein biomarkers. 2) Our analysis was limited to the COPDGene study. In the future, such analyses could be conducted in other ongoing studies collecting CT scan and omics data in populations enriched for COPD.
In summary, deep learning applied to CT images, and transcriptomic biomarkers in COPD identified two main inflammatory processes related to CT image features that can be broadly defined as emphysema and airway disease. The emphysema-related process was most enriched for pathways related to neutrophilic inflammation. The airway axis differed from previously reported disease axes learned from phenotypic data alone and it was negatively correlated with emphysema. Finally, there was also a strong relationship between the core-peel distribution of emphysema and systemic inflammation. In the future, these integrative machine learning methods can be refined for more fine-grained interpretability and extended to include other sources of biological information.
Data Availability
All data produced are available online at dbGaP
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000179.v6.p2
Supplemental Methods
Subject Enrollment and Data Collection
COPDGene enrolled 10,198 subjects with a minimum 10 pack-years lifetime smoking history at 21 centers across the United States (NCT00608764, www.copdgene.org)(1). Five-year follow-up data are available for 6,717 subjects, and 10-year follow-up visits are currently being completed. Subjects underwent spirometry, questionnaire assessments, standardized inspiratory and expiratory chest CT imaging, and genome-wide genotyping. Starting at the second visit (year 5), PAXgene blood RNA tubes were collected. Each center obtained institutional review board approval, and all participants provided written informed consent.
We used the following cross-sectional covariates from the COPDGene second study visit in our analysis – age, race, gender, postbronchodilator spirometric measures, Body Mass Index (BMI), St. George’s Respiratory Questionnaire (SGRQ) total score, modified Medical Research Council (mMRC) Dyspnea Scale, and the 6-minute walk distance The variable “frequent exacerbator (history)” indicates whether the subject had >=2 self-reported exacerbations during the 12 months before the second visit. Exacerbations were defined as worsening respiratory symptoms treated with systemic steroids and/or antibiotics.
The following quantitative CT measurements were analyzed: Perc15 (15th percentile value of Hounsfield unit (HU) in the lung region), % emphysema (the percentage of low attenuation areas less than -950 HU in the inspiratory CT scan), % air trapping (the percentage of low attenuation areas less than -856 HU in the expiratory CT scan), Pi10 (The square root of the wall area of a theoretical airway with an internal lumen perimeter of 10 mm) and %WA segmental (the percentage of airway wall area for 3rd generation bronchi). The variable  is defined as 100 times the logarithm of the ratio of perc15 value in the peel to the core region of the lung. We define the peel region as the lung region that is not more than 5mm from the boundaries of the lung and the core region to be more than 20mm from the lung boundary. A larger value of
is defined as 100 times the logarithm of the ratio of perc15 value in the peel to the core region of the lung. We define the peel region as the lung region that is not more than 5mm from the boundaries of the lung and the core region to be more than 20mm from the lung boundary. A larger value of 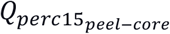 represents that more emphysema is in the peel area of the lung.
represents that more emphysema is in the peel area of the lung.
Using data gathered from a subset of subjects who had already completed the third (10-year) COPDGene Study visit, we included variables on prospective change in lung function and exacerbations in our analysis. ΔFEV1 %predicted and ΔFEV1/FVC are computed by subtracting the visit 3 value from the visit 2 value of FEV1 or FEV1/FVC and dividing it by the number of years between the two visits. We used “frequent exacerbator (future)” to represent whether the subject had at least two self-reported exacerbations during the 12 months prior to the third study visit.
Extracting CSRL Features from CT Scans
We resampled inspiratory CT scans to isotropic 1mm3 resolution. The Hounsfield Units (HU) were mapped to the intensity window of [−1024, 240]. We selected the CT scan from a healthy subject as the anatomical atlas. Next, we divided the atlas image into non-overlapping patches with the size 323 mm3 using the sliding window approach, which resulted in 581 patches. The center locations of these patches were used as anatomical landmarks. Then we performed affine registration using ANTs (2) to map these atlas landmark coordinates to every CT scan, which maintained the anatomical correspondences between images. Finally, we divided each CT scan into 581 patches centering at the mapped anatomical landmarks, with a size of 323 mm3. We extracted the features for each patch using the context-aware self-supervised representation learning (CSRL) model (3). The CSRL model was pre-trained using the inspiratory CT scans collected during the first COPDGene study visit. This model embedded each patch in 128-dimensional space, resulting in 128 CSRL features, assuming the embedding of similar samples are closer and diverse samples are far from each other.
RNA-seq Data Generation and pre-processing
Total blood RNA was collected at Visit 2 in PAXgene TM Blood RNA tubes from the Qiagen PreAnalytiX PAXgene Blood miRNA Kit (Qiagen, Valencia, CA). Paired-end reads were generated from Illumina sequencers. Gene transfer format (GTF) annotation was downloaded from the Biomart Ensembl database (Ensembl Genes release 94, GRCh38.p12 assembly).
Gene counts were derived from Salmon isoform estimates summarized at the gene level using the tximport package. The gene and isoform count data used for this analysis are available in the Gene Expression Omnibus(4) (accession number GSE158699).
Genomic features with very low expression (average counts per million (CPM) < 0.2 or number of subjects with CPM < 0.5 was < 50) or extremely highly expressed genes (number of subjects with CPM > 50,000 was less than 50) were filtered out prior to applying trimmed mean of M values normalization from edgeR (v3.24.3), which accounts for differences in sequencing depth(5). Counts were also transformed to log2 CPM values and quantile-normalized to further remove systematic noise from the data.
Learning Image-Expression Axes (IEAs)
To select the most relevant genes to use in our machine learning modeling, we first concatenated 128 CSRL features in 581 patches for each subject into a 581× 128 vector and conducted Principal Component Analysis (PCA) to reduce the dimensionality to 128. Then we trained a linear regression model with these 128 Principal Components (PCs) to predict each gene expression level and computed the p-value for each regression model using an F-test. We corrected for multiple comparisons using the Benjamini–Hochberg procedure(6), and for the final model, we included genes with an adjusted p-value <0.01 based on sensitivity analyses described below. Gene expression data were centered and scaled prior to analysis (mean 0 and unit variance).
We implemented a machine learning model in PyTorch (v1.10.0) to extract Image-Expression Axes (IEAs) for each subject, utilizing the CSRL features of 581 patches for each subject. We first fed CSRL features to a multilayer perceptron (MLP) to obtain a low-dimensional representation for each patch. Then we obtained the subject-level disease axes, which we call Image-Expression Axes (IEAs), by summarizing the 581 sets of low-dimensional patch-level representations using a Product of Expert (PoE) model(7). The PoE model assumes the probability density functions (PDF) of the subject-level disease axes are given by the product of PDFs of the patch-level representations. We apply independent constraints for the IEA with the Hilbert-Schmidt independence criterion (HSIC) (8) to ensure statistical independence of each identified axis with the goal of each axis capturing an independent disease process. We added another linear layer that took the IEAs as the input and outputted the estimated expression levels of all selected genes simultaneously. We define the objective function of the model to minimize the mean-squared errors between the estimated and the observed expression levels of the selected genes. The parameters of the model were optimized via Adam(9). We include the details of the deep learning method in the next section.
The Deep Learning Model
The context-aware self-supervised representation learning (CSRL) extracted 128 features for each of the 581 patches. We denote these CSRL features using a vector xn,p ∈ R128, where n ∈ {1, …, M}, and p ∈ {1, …, 581} represent the indices for subjects and patches, respectively, and M is the number of subjects in the training set. We first learn an L-dimensional patch-level representation for each patch rn,p ∈ RL. We assume that each element of rn,p follows a Gaussian distribution. We assume that the prior distribution for each element of rn,p is given by the standard Gaussian distribution with zero mean and unit variance such that p(rn,p,l) = N (0, 1), and its posterior can be approximated by  , where l ∈ {1, …, L} is the index for the representation. The mean and variance of the Gaussian distribution q(rn,p,l), denoted as μl(·) and
, where l ∈ {1, …, L} is the index for the representation. The mean and variance of the Gaussian distribution q(rn,p,l), denoted as μl(·) and  are the output of Multi-Layer Perceptron (MLP) with 3 hidden layers. We let the three hidden layers of the MLP contain 96, 64 and 48 nodes, respectively, with Rectified Linear Unit (ReLU) activation functions. We use θ to represent all the parameters for this MLP.
are the output of Multi-Layer Perceptron (MLP) with 3 hidden layers. We let the three hidden layers of the MLP contain 96, 64 and 48 nodes, respectively, with Rectified Linear Unit (ReLU) activation functions. We use θ to represent all the parameters for this MLP.
We summarize the patch-level representations into a subject-level IEA zn ∈ RL with a product-of-expert model(7), assuming that the distribution of each dimension in zn is given by  . Since zn,l is a produce of Gaussian distributions, zn,l also follows a Gaussian distribution, such that
. Since zn,l is a produce of Gaussian distributions, zn,l also follows a Gaussian distribution, such that 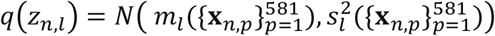 , where
, where
 Note that both ml(·) and
Note that both ml(·) and  are functions of
are functions of  , and parameterized by the MLP parameters θ.
, and parameterized by the MLP parameters θ.
We assume that each dimension in zn is independent with each other, such that each zn,l represents an independent disease process of COPD. Therefore, we introduce a pairwise Hilbert-Schmidt independence criterion (HSIC) penalty in the objective function, such that zn,i and zn,j are statistically independent for all i, j ∈ {1, …, L}, denoted by  ,where HSIC is estimated as described in Section 5.2 in (8).
,where HSIC is estimated as described in Section 5.2 in (8).
We denote the gene expression levels for each subject is given as yn ∈ RD, where D represents the number of genes. We estimate yn with a linear layer using zn as the input, such that the estimator is given as ŷn = Wzn, where W ∈ RD×L represent the parameters in the linear model. We define the objective function of the model to minimize the mean-squared errors between the estimated and the observed expression levels of the selected genes.
We optimize the evidence lower bound (ELBO)(10) as the objective function, which can be given as
 where
where 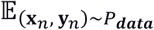 represent the expected values are estimated with the observed data, and DKL(· || ·) represents the Kullback–Leibler. The hyper-parameters β and λ control the trade-off between the contributions of different terms in the objective function. In the primary experiments we let β = 0.1 and λ = 1.
represent the expected values are estimated with the observed data, and DKL(· || ·) represents the Kullback–Leibler. The hyper-parameters β and λ control the trade-off between the contributions of different terms in the objective function. In the primary experiments we let β = 0.1 and λ = 1.
We minimized the objective function utilizing Adaptive Moment Estimation (Adam) (9). In the optimization, we utilized the reparameterization trick, i.e., we let  for each n ∈ {1, … N} and l ∈ {1, … L}, where ∈ is a random sample from 𝒩(0, 1).
for each n ∈ {1, … N} and l ∈ {1, … L}, where ∈ is a random sample from 𝒩(0, 1).
Model training and Validation
We analyzed the inspiratory CT scans and the RNA-seq data of 1,223 subjects. We randomly split the data into training and testing sets containing 923 and 300 subjects, respectively. Model training was performed in the training set of 923 subjects using five-fold cross-validation, giving us five models. We aligned the IEAs by pairing the IEA from different models, such that the Pearson correlation coefficient is maximized. We standardize the aligned IEAs of each model, such that they have zero mean and unit variance. The final IEAs were given by taking the average value of the aligned IEAs given by five models.
We first validated our model by examining whether the learned IEAs were reproducible if different subsets of samples were used for training during the 5-fold cross-validation. We quantified the reproducibility via computing the mean and standard deviations of the Pearson correlation coefficients between the aligned axes. Then, we examined whether the IEAs were reproducible if different subsets of genes were used for training. To do so, we varied the thresholds for the gene selection process such that different subsets of genes were selected, conducted a 5-fold cross-validation, and obtained the final IEA by taking the average value of the standardized aligned IEAs of 5 folds. We compared these final IEAs trained with different subsets of genes by computing the Pearson correlation coefficients.
Association of IEAs with Clinical Measurements
To understand the association between the IEAs and the clinical measurements, we conducted a univariable analysis by computing Pearson correlation coefficients between each of the IEAs and each of the clinical measurements. We conducted multivariable analysis via training an Ordinary Least Squares (OLS) model for continuous measurements and training a logistic regression model for categorical measurements. We conducted a survival analysis with the Cox proportional hazards model(11). In these multivariable analyses, we adjusted for the following covariates: age, race, gender, pack-years of smoking, current smoking status for all models. To determine whether the IEAs improved the prediction of each outcome, we compare the models with both IEAs as predictors to a reduced model without the IEAs using likelihood-ratio tests.
In this study, we utilize  to measure the peel-core distribution of emphysema. To better understand how the peel-core distribution of emphysema is related to the IEAs, we conducted sensitivity analyses where we separated the lung into bands according to the distance to the lung boundary. We compute logarithm the ratio of perc15 value in different bands to the core region of the lung and observe how it is correlated to the IEAs.
to measure the peel-core distribution of emphysema. To better understand how the peel-core distribution of emphysema is related to the IEAs, we conducted sensitivity analyses where we separated the lung into bands according to the distance to the lung boundary. We compute logarithm the ratio of perc15 value in different bands to the core region of the lung and observe how it is correlated to the IEAs.
To compare IEAs to previously identified COPD subtypes using k-means (12), we investigated whether the IEA values are different across the identified subtypes with the student’s t-test.
Comparison of IEAs to Other COPD Subtypes and Disease Axes
We compared the IEAs to the following disease axes obtained with machine learning approaches: 1. COPD Factor Analysis Axes (FAs): Previously published phenotype disease axes identified through factor analysis of quantitative CT measurements and pulmonary function variables(13). 2. PCA Image Only Axes (PCA-I): Disease axes are constructed from imaging data only by applying Principal Component Analysis (PCA) to the same CSRL features used in the IEA analysis, but without using the gene expression data. Since the FAs were available only at the COPDGene first study visit, we generated IEAs from the CT scan data at visit 1 in order to directly compare FAs and IEAs. The comparative analyses were: 1. Calculating the Pearson correlation coefficients between IEAs and other disease axes; 2. Association analyses of FAs and PCA-I disease axes to the same cross-sectional and longitudinal clinical measurements described above; 3. likelihood-ratio tests to compare models utilizing both IEAs and FAs or PCA-I axes to baseline models including either FAs or PCA-I without IEAs.
To understand whether the IEAs capture gene-relevant information, we quantified the proportion of variation of gene expression explained by the IEAs in the COPDGene visit 2 data. We compared this to the proportion of expression variance explained by the PCs of CSRL features (PCA-I) to understand whether the IEAs capture more gene-relevant information.
Differential Expression and Usage Analyses
To identify the genes and pathways associated with each of the IEAs identified in this study, we performed differential gene expression using the limma R package (v3.38.3)(14,15). We adjusted for the following covariates: age, race, gender, pack-years of smoking, current smoking status, forced expiratory volume in one second (FEV1), CBC cell count proportions, library prep batch, and CT scanner model. Multiple comparisons were corrected with the Benjamini-Hochberg method to control the false discovery rate (FDR) at 10%(6). Gene ontology (GO) pathway enrichment analysis was performed using the GO “Biological Process” gene sets with p-values calculated with the Fisher exact test statistic using the weight01 algorithm in topGO (v2.33.1) (16) that accounts for dependency in GO topology(17). We reported GO pathways with at least three significant genes, and the threshold for statistical significance was an adjusted P-value < 0.001.
Supplemental Results
As shown in Error! Reference source not found., both IEAs were negatively associated with emphysema peel/core distribution 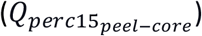 , indicating that higher IEA values are associated with more emphysema in the core/central regions of the lung. To explore this association further and to determine whether this may be driven by artifacts of lung segmentation, we performed a sensitivity analysis where the lung was separated into concentric bands according to the distance to the lung boundary, as shown in Supplemental Figure E3. While the core region was always defined as the lung regions >20 mm from the lung boundary, the peel region was alternatively defined using each of the concentric bands between the core and lung periphery. For the IEAemph axis, the negative correlation to
, indicating that higher IEA values are associated with more emphysema in the core/central regions of the lung. To explore this association further and to determine whether this may be driven by artifacts of lung segmentation, we performed a sensitivity analysis where the lung was separated into concentric bands according to the distance to the lung boundary, as shown in Supplemental Figure E3. While the core region was always defined as the lung regions >20 mm from the lung boundary, the peel region was alternatively defined using each of the concentric bands between the core and lung periphery. For the IEAemph axis, the negative correlation to 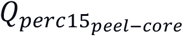 was observed for all concentric bands outside the core; however, the IEAairway axis was significantly negatively correlated to peel-core emphysema only when the peel was defined as the outermost band (Supplemental Table E18). This suggests that IEAemph reflects a consistently positive relationship with core emphysema, whereas, for IEAairway, it is not currently clear whether this reflects a real biological phenomenon at the extreme periphery of the lung, or this may be due to artifacts of segmentation.
was observed for all concentric bands outside the core; however, the IEAairway axis was significantly negatively correlated to peel-core emphysema only when the peel was defined as the outermost band (Supplemental Table E18). This suggests that IEAemph reflects a consistently positive relationship with core emphysema, whereas, for IEAairway, it is not currently clear whether this reflects a real biological phenomenon at the extreme periphery of the lung, or this may be due to artifacts of segmentation.
Supplemental Figures and Tables
The following tables are included in the supplemental excel file:
Table E1. Cross-validation performance in IEA training.
Table E2. Linear Regression with image-expression axes (IEAs) and COPD measurements with COPDGene visit 2 data.
Table E3. Logistic regression and Cox proportional harzard models with image-expression axes (IEAs) and COPD measurements with COPDGene visit 2 data.
Table E4. Pearson’s correlation between image-expression axes (IEAs) and COPD-related characteristics and health outcomes, measured on 1,527 subjects from another subset of the COPDGene dataset that had not been used for model training.
Table E5. Linear Regression with image-expression axes (IEAs) and COPD measurements with 1,527 subjects from another subset of the COPDGene dataset that had not been used for model training.
Table E6. Logistic regression and Cox proportional harzard models with image-expression axes (IEAs) and COPD measurements with 1,527 subjects from another subset of the COPDGene dataset that had not been used for model training.
Table E7. Characteristics of subgroups defined by dividng the Image-Expression Axes (IEAs) into quadrants.
Table E8. Covariances between Image-expression Axes (IEAs), factor analysis axes (FAs), and PCA image-only axes (PCA-Is) on COPDGene visit 1 data.
Table E9. Pearson’s correlation coefficient between image-expression axes (IEAs), factor analysis axes (FAs), PCA image only axes (PCA-Is), and health outcomes on COPDGene visit 1 data.
Table E10. Linear regression analysis with image-expression axes (IEAs) and factor analysis axes (FAs) on COPDGene visit 1 data.
Table E11. Logistic regression and Cox model with image-expression axes (IEAs) and factor analysis axes (FAs) on COPDGene visit 1 data.
Table E12. Linear regression analysis with image-expression axes (IEAs) and PCA Image Only Axes (PCA-Is) on COPDGene visit 1 data.
Table E13. Logistic regression and Cox model with image-expression axes (IEAs) and PCA Image Only Axes (PCA-Is) on COPDGene visit 1 data.
Table E14. Differentially Expressed Genes (DEGs) associated with the IEAemph (adjusted p-value <.10).
Table E15. Differentially Expressed Genes (DEGs) associated with the IEAairway (adjusted p-value <.10).
Table E16. Significant Gene Ontology enrichment terms for IEAemph (adjusted p-value < 0.001).
Table E17. Significant Gene Ontology enrichment terms for IEAairway (adjusted p-value < 0.001).
Table E18. The correlation between perc15 ratio and IEAs.

Consort Diagram showing the subjects used in the analysis.

Data flow for model training and validation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lung “bands” used in the peel-core sensitivity analysis. To determine the robustness of the IEA association to peel-core emphysema, the 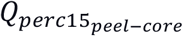 variable was recomputed using a series of lung bands defined based on the distance to the lung boundary. R is defined as the distance to the boundaries of the lung.
variable was recomputed using a series of lung bands defined based on the distance to the lung boundary. R is defined as the distance to the boundaries of the lung.
Footnotes
Sources of Support: This work was supported by NHLBI K08 HL141601, R01 HL124233, R01 HL126596, R01 HL147326, U01 HL089897, and U01 HL089856. The COPDGene study (NCT00608764) is also supported by the COPD Foundation through contributions made to an Industry Advisory Committee comprised of AstraZeneca, Bayer Pharmaceuticals, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer and Sunovion.
Competing Interests: PJC has received grant support from Bayer and consulting fees from Novartis and GSK. CPH reports grant support from Bayer, Boehringer-Ingelheim and Vertex, and consulting fees from AstraZeneca and Takeda. EKS has received grant support from Bayer and GSK.
Replicate the results on 1,527 subjects from another subset of the COPDGene dataset that had not been used for model training.
References
References



