
Abstract
A well-functioning placenta is essential for fetal and maternal health throughout pregnancy. Using placental weight after term delivery as a proxy for placental growth, we report genome-wide association analyses in the fetal (n = 65,405), maternal (n = 61,228), and paternal (n = 52,392) genomes, yielding 40 independent association signals. Twenty-six signals are confidently classified as fetal only, four maternal only, and three fetal and maternal. A maternal parent-of-origin effect is seen near KCNQ1. Genetic correlation and colocalization analyses reveal overlap with birth weight genetics, but twelve loci are classified as predominantly or only affecting placental weight, with connections to placental development and morphology, and transport of antibodies and amino acids. Mendelian randomization analyses indicate that fetal genetically mediated higher placental weight is causally associated with risk of preeclampsia or shorter gestational duration. Moreover, these analyses support a role for insulin produced by the fetus in regulating the growth of the placenta, providing a key link between fetal and placental growth.
Main
The placenta is a remarkable pregnancy organ formed after implantation of the blastocyst into the uterine wall. The intimate placental connection between mother and fetus provides nutrients and oxygen to the developing fetus, while removing waste products from the fetal blood. The placenta produces hormones, growth factors, and cytokines, and allows maternal IgG antibodies to be passed to the fetus, giving newborns innate immunity. Suboptimal placentation can lead to intrauterine growth restriction1, spontaneous abortion, preterm birth2, and preeclampsia3,4, and a poorly functioning placenta is associated with higher risk of growth restriction5, adverse neurodevelopment6, and cardiometabolic diseases7–11.
Placental weight (PW) is an easily available measure often used in epidemiological studies12,13 as a proxy for placental growth and function. The close relationship between placental and fetal growth is reflected by a positive correlation (r = 0.6) between PW and birth weight (BW) in large cohorts12,14. Several genome-wide association studies (GWAS) have identified genetic loci in the maternal and fetal genomes associated with BW15,16, and BW–associated loci are enriched for placental expression quantitative trait loci (eQTLs)17. However, no GWAS of PW is available to date, and it remains unclear how the genetics of placental growth relate to the genetics of fetal growth or to the genetics of adverse pregnancy outcomes such as preeclampsia. While the placenta is primarily composed of cells with fetal origin, it is intricately connected to maternal tissues and circulation18,19. Genetic analyses offer the opportunity for insight into the complex interplay of direct fetal, indirect maternal, and parent-of-origin effects (POE), which we hypothesize underlie placental growth and function.
We conducted the first GWAS of PW in term, singleton pregnancies, meta-analyzing data from 28 European studies. Analyses of 19,861 child-mother-father trios with PW measurements enabled a better understanding of the relationship between fetal and maternal effects, including POE. We also categorized loci according to their association with BW, examined genetic links between PW and pregnancy, perinatal and later-life outcomes, and used Mendelian randomization (MR) to assess causal relationships between maternal and offspring characteristics and PW.
Results
Meta-analyses of fetal, maternal, and paternal GWASs
We performed GWAS meta-analyses of PW adjusted for fetal sex and gestational duration against the fetal (n = 65,405), maternal (n = 61,228), and paternal genomes (n = 52,392) (Fig. 1.). Methods and Supplementary Tables 1-6 provide information on data collection and genotyping. After data cleaning and imputation, 11 million SNPs were analyzed. The fetal GWAS meta-analysis identified 32 independent loci at P < 5 × 10−8, the maternal analysis identified four loci, and the paternal identified two loci (Fig. 2, Table 1, Supplementary Table 7). Approximate conditional and joint analysis (COJO) further identified secondary association signals at three fetal loci (Methods; Table 1; Supplementary Table 7). A comparison of effect sizes against minor allele frequencies for those 41 association signals was in line with expectations from statistical power (Supplementary Fig. 1). We also conducted analyses adjusted only for sex (i.e. not gestational age), which showed high genetic correlations with our main analysis results (fetal r_g = 1.00, SE = 0.001, P ≤ 1 × 10−16; maternal r_g = 0.99, SE = 0.001, P ≤ 1 × 10−16; paternal r_g = 1.00, SE = 0.004, P ≤ 1 × 10−16). Four additional loci reached genome-wide significance in the fetal sex-adjusted analyses, all of which were close to genome-wide significance in our main fetal analysis (Supplementary Table 8). Regional association plots for all genome-wide significant loci in the main analyses are shown in Supplementary Fig. 2a-e.

Abbreviations: eQTL, expression quantitative trait loci; GWAS, genome-wide association study; HRC, Haplotype Reference Consortium; MAC, minor allele content; meQTL: methylation quantitative trait loci; PC, principal component.

Manhattan plots of –log10 P values across the chromosomes and corresponding quantile–quantile plot of observed versus expected –log10 P values for meta-analyses of SNP associations with placental weight in a, the fetal GWAS; b, the maternal GWAS; and c, the paternal GWAS.
Resolving fetal and parental contributions to association signals
Fetal genotype is correlated with both maternal and paternal genotype (each r = 0.5). While the fetal genotype may influence PW directly, the maternal genotype itself may have effect via the intrauterine environment. To calculate the SNP heritability of the maternal, paternal, and offspring contributions we used a framework within genomic structural equation modelling (SEM)20 developed by Moen et al.21 (Methods). We found evidence for substantial contributions from the fetal (h2=0.22 (SE=0.03)) and maternal (h2=0.12 (SE=0.02)) genomes to variation in placental weight, and a small component from the paternal genome (h2=0.06 (SE=0.02))(Supplementary Table 9, Supplementary Fig. 3). We also found genetic correlation between the three latent variables suggesting that the fetal effect on placental weight was negatively correlated with both maternal and paternal effects, while maternal and paternal effects on placental weight were positively correlated (Supplementary Table 9).
To estimate fetal-specific, maternal-specific, or paternal-specific effects on PW at each identified locus, we first applied a weighted linear model (WLM)15 to the GWAS meta-analysis summary statistics. The WLM can accurately approximate conditional effects (e.g. fetal effect conditional on any maternal effect, or vice versa) in absence of genotyped mother-child pairs by incorporating results from all samples, even those overlapping between the three GWAS meta-analyses (Methods), to maximize statistical power. We applied formulations of the WLM allowing for non-zero paternal effects, or fixing the paternal effects to zero (Supplementary Table 10). At the 41 association signals, we applied the classification method used for BW15 (Methods) to categorize the SNPs as having fetal or maternal effects. Hence, 26 SNPs were classified as fetal only, four as maternal only, two as fetal and maternal with effects in opposite directions, and one as fetal and maternal with effects in the same direction. We could not resolve the classification for the remaining eight loci (Supplementary Table 10, Supplementary Fig. 4a, Supplementary Fig.4b).
We complemented the WLM analysis with within-family analyses in the child-mother-father trio subset from the Norwegian Mother, Father, and Child Cohort (MoBa)22. First, the associations of fetal and parental genotypes with PW were conditioned against each other (n = 19,861 independent trios, Supplementary Table 10). There was good agreement between mode of association categories based on WLM results and those from conditional analyses in trios. Taking advantage of the availability of phased genotypes for MoBa children, we further decomposed the association signals into their mode of transmission (i.e., maternal transmitted, maternal non-transmitted, paternal transmitted, and paternal non-transmitted alleles). Since several loci were already known to be associated with BW (see below), we compared our results with a recent analysis of BW-mode of transmission16 (Supplementary Table 11). For loci associated with both traits, effect size estimates and mode of transmission were consistent between BW and PW analyses (Fig. 3a, Supplementary Fig. 5). Among the eight loci that remained unclassified after WLM analysis of PW, five were identified previously in BW GWAS and classified as fetal16, and one was classified as fetal and maternal with same direction of effect (Supplementary Table 11).

a, Effect size estimates divided by standard error in the mode of association analyses for the 41 association signals and the association of the maternal non-transmitted, maternal transmitted, paternal transmitted, and paternal non-transmitted alleles with standardized PW and BW adjusted for sex and gestational duration in MoBa, and with BW according to Juliusdottir et al.16 (a blank value indicates that no proxy was found with r2 ≥ 0.2). b, Effect size estimates for standardized PW and BW adjusted for sex and gestational duration for rs2237892 near KCNQ1 in MoBa, thin and thick error bars respectively represent 95% confidence intervals and one standard error on each side of the effect size estimate. c,d, Regional plots at the KCNQ1 locus, plotting the negative base 10 logarithm of the unadjusted P-value against genomic location, for the maternal non-transmitted, maternal transmitted, paternal transmitted, and paternal non-transmitted alleles in their association with standardized placental (c) and BW (d) adjusted for sex and gestational duration in MoBa. The leading variants for PW and BW, rs2237892 and rs234864, are annotated with a diamond and a circle, respectively. Points are colored according to their LD with rs2237892. Abbreviations: MnT, maternal non-transmitted alleles; MT, maternal transmitted alleles; PT, paternal transmitted alleles; PnT, paternal non-transmitted alleles.
For the signal identified in the paternal GWAS near EBF1 (rs75512885), our sample size did not allow resolving the mode of association using WLM or parent-offspring trio analyses, but this variant is in moderate LD with rs72813918 (r2 = 0.57), classified previously as fetal for BW16 (Supplementary Table 11). Colocalization analysis suggested that this signal for BW was independent of the nearby fetal association signal for PW (rs67265526; posterior probability for independent signals > 0.99). The lead SNP of the other paternal GWAS locus (rs2207099, near LOC339593) colocalized with the nearby fetal lead SNP (rs6040436; posterior probability for shared association = 0.97) (Supplementary Table 7), and was therefore excluded from subsequent analyses, leaving 40 independent association signals.
For all loci, we inspected the distance of the lead SNP to known imprinted genes according to www.geneimprint.com (Supplementary Table 11). Only one lead variant was found near imprinted genes; rs2237892 located in intron 10 of the imprinted gene KCNQ1. Indeed, the mode of transmission analysis in MoBa showed that the association with PW was only conferred by the maternally transmitted allele (Fig. 3b, Supplementary Fig. 5) consistent with the known maternal-only expression of KCNQ1 and nearby CDKN1 genes. This variant shows POE on type 2 diabetes risk23, where the maternally inherited risk allele (C) corresponds to the maternally inherited PW-decreasing allele identified here. Another nearby variant, rs234864, has been reported16 as a maternally-transmitted effect only for BW. The very low LD (r2 = 0.05) and very different minor allele frequencies (5.6% vs. 49.4%) between the two variants suggest that these may be independent signals, both imprinted (Fig. 3c). While the maternally transmitted allele of the BW variant rs234864 was strongly associated with both PW and BW in MoBa, the maternally transmitted allele of the PW variant rs2237892 showed directionally consistent, but weaker evidence of association with BW in MoBa (Fig. 3d). This is in agreement with the lower fetal effect size detected of rs234864 on BW in previous BW GWAS16 compared with the effect on PW (Supplementary Table 11).
Correlations between PW and BW
The strong phenotypic correlation reported between PW and BW12,14 was confirmed in the MoBa samples in this study (Spearman’s r = 0.59, adjusted for sex and gestational duration). We applied LD score regression24 to estimate genetic correlations with published BW association summary statistics15, analyzing both main GWAS summary statistics, and WLM-adjusted estimates of the conditional fetal, maternal and paternal effects (Fig. 4). Using WLM-adjusted effects for both PW and BW, the fetal and maternal genetic correlations between PW and BW remained strong. We observed a negative genetic correlation between fetal-specific effects on BW and maternal-specific effects on PW (Fig. 4c), whereas fetal-specific effects on PW showed no genetic correlation with maternal-specific effects on BW (Fig. 4d). There was no genetic correlation between paternal-specific effects on PW and either maternal- or fetal-specific effects on BW after WLM-adjustment.

a, Genetic correlation estimates for PW (current study - illustrated by circles or triangles with 95% CI) and BW (using 4 types of estimate) from Warrington et al.15. “Own birth weight” means fetal GWAS of birthweight, and “offspring birth weight” means maternal GWAS of offspring birth weight. Values are provided for fetal, maternal, and paternal effects before and after WLM adjustment. Error bars represent 95% confidence intervals. Abbreviations: CI, confidence interval; Rg, genetic correlation; WLM, weighted linear model.
From the largest available GWAS of BW16, we selected BW loci within 500 kb of PW loci (Supplementary Table 7). Twenty-eight of the 40 independent PW signals were also reported for BW or had a BW lead SNP nearby. All these were within 100 kb of the PW lead SNP except rs876987 at SLC7A5 (> 419 kb). Colocalization analysis suggested that 19 of these represent a shared underlying association (posterior probability for shared signal > 0.8), five signals were distinct (posterior probability for separate signals > 0.8), and the final four loci were uncertain (both posterior probabilities < 0.8).
Given the large proportion of signals colocalizing with BW loci, we aimed to classify loci showing evidence of associations with both PW and BW from those showing only (or predominantly) evidence of association with PW, by comparing PW and BW effect estimates (Supplementary Table 7, Methods). Twelve signals were classified as only or predominantly PW, and the remaining 28 were classified as both PW and BW signals, with one of these (near SLC7A5) showing opposite directions of effect both with P < 0.05. The results obtained from WLM-adjusted analysis were largely consistent (Fig. 5, Supplementary Fig. 6, Supplementary Table 7). Thus, 12 of the 40 independent signals only influence PW, or have effects on PW that are larger than any estimated effect on BW (Table 1).

Scatter plots comparing effect size estimates from the PW GWAS with those from the BW GWAS15. The left column shows fetal genome associations, and the right shows maternal. The top row shows SNPs classified as fetal only effects on PW (Supplementary Table 7). The middle row shows SNPs classified as maternal, or maternal and fetal, and the bottom row shows unclassified SNPs. SNPs with minor allele frequency below 5% have been excluded to improve resolution. The full figure including the lower frequency SNPs is shown in Supplementary Fig. 6. Colors indicate the classifications, which are given in the key below the figure. Abbreviations: BW, birth weight; GWAS, genome-wide association study; PW, placental weight. Error bars represent 95% confidence intervals.
Connections to placental development and morphology, and transport of antibodies and amino acids
To prioritize key tissues and cell types relevant to fetal genes implicated by our identified loci, we selected the 31 protein-coding genes closest to the index SNP of the 32 loci identified in the fetal GWAS (i.e. excluding LOC339593; Table 1) and examined their expression using tissue-specific mRNA abundance data from the Human Protein Atlas and a scRNA-seq data set of over 70,000 single cells at the decidual-placental interface in early pregnancy (Methods). Among 61 tissues, the 31 genes ranked higher in terms of mRNA abundance than other genes in placenta (P = 1.8 × 10−4) (Supplementary Fig. 7, Supplementary Table 12). In the single cell analysis among the 32 different cell types at the early maternal–fetal interface, expression of the 31 genes ranked significantly higher than other genes in cell types of fetal origin, including fetal endothelial cells (P = 6.2 × 10−5) and syncytiotrophoblasts (P = 1.9 × 10−4), but they also ranked higher in maternal innate lymphocyte cells (P = 1.2 × 10−3) (Supplementary Fig. 8, Supplementary Table 12).
To further investigate associations between PW loci and gene expression or methylation in placental tissue, we queried lead PW SNPs (and their proxies r2 ≥ 0.8) against eQTL data from placenta in the Rhode Island Child Health Study dataset25. Lead PW SNPs tagged placental eQTL at four loci (HSPA4, TBX20, SLC7A5, JAG1) (Supplementary Table 7). We additionally investigated whether any of the 40 independent lead SNPs were associated with placental DNA methylation at CpG sites within 0.5 Mb. Using Gen3G data26,27, we identified placental meQTLs at 21 of the 40 independent signals (FDR ≤ 0.05 extrapolated from the genome-wide beta distribution by TensorQTL; Supplementary Fig. 8). Among the 21 lead SNPs with identified meQTLs, individual SNPs were associated with placental DNA methylation at up to 15 CpG sites (Supplementary Fig. 9). Supplementary Table 7 shows meQTLs with lowest P-value for each lead SNP.
We combined the placental expression and methylation data with further look-ups of lead PW SNPs in available GWAS of gene expression, plasma protein levels, diseases and traits (Supplementary Tables 7, 13, 14, 15). These searches implicated several candidate genes and potential biological insights. For example, the PW-raising allele of rs723177 at the fetal RPL31P11 locus (Supplementary Fig. 2a) is an eQTL and pQTL for FCGR2A and FCGR2B (receptors for the Fc region of IgG complexes) and associates with higher plasma protein levels (Supplementary Tables 14, 15), suggesting a potential role in maternal antibody transfer across the placenta28. The lead variant rs723177 is also the strongest placental meQTL in the region at the CpG site cg27514565 (overlapping DNase I Hypersensitive Sites [DHS] for primitive/embryonic and myeloid/erythroid tissues) located between FCGR2C and FCGR3B, and two meQTL (at cg14354529 and cg15531363) overlapping with trophoblast-specific DHS located downstream of FCRLA and FCRLB (Supplementary Fig. 6). Associations at lead SNP rs10486660, at another fetal locus, suggest a role for this locus in placental morphology: the SNP is intragenic in the T-box transcription factor gene, TBX20 (Supplementary Fig. 2a) and marks an eQTL for TBX20 in placenta (Supplementary Table 7). Approximately 50% of TBX20 placental expression is found in trophoblast cells29, and TBX20-null mice are likely to show abnormal placental morphology compared to wild-type mice30. Third, the lead SNP rs112635299 is in LD (r2 = 1.0) with rs28929474, a missense variant in the serpin peptidase inhibitor gene SERPINA1 (Supplementary Fig. 2a). The SNP is associated with several traits, including levels of >20 circulating plasma proteins31 (Supplementary Table 15) and the cause of autosomal recessive alpha-1 antitrypsin deficiency32. Within the placenta, SERPINA1 expression regulates expression of inflammatory cytokines and serine protease HTRA1-induced trophoblast invasion through induction of endoplasmic reticulum stress31. Finally, lead fetal SNP, rs876987, being the only variant showing an inverse effect on PW relative to BW, is intragenic in SLC7A5, which encodes solute carrier family 7 member 5 (Supplementary Fig. 2a). The heterodimer encoded by SLC7A5 and SLC3A2 is a sodium-independent, high-affinity amino acid exchanger over membranes of several organs and is responsible for uptake of essential amino acids in placenta. Lead variant rs876987 is an eQTL for SLC7A5 in placenta and a meQTL for 15 CpG sites, with the strongest association detected at cg03408354 (P = 4.7 × 10−49; located in intron 7 of SLC7A5) (Supplementary Table 7; Supplementary Fig. 9d).
PW and pregnancy complications including preeclampsia
We tested whether the 40 independent lead SNPs were associated with pregnancy and perinatal traits, using published GWAS summary statistics for nausea and vomiting of pregnancy or hyperemesis gravidarum33, preeclampsia34, gestational duration35, miscarriage (recurrent and spontaneous)36, and ten cytokines assayed from neonatal blood spots37. The SNPs showed more associations with nausea and vomiting of pregnancy (maternal genotype effects), and with gestational duration, preeclampsia, and neonatal IgA levels (all fetal genotype effects) than expected under the null distribution (Supplementary Fig. 10, Supplementary Table 16)35,36. Scatter plots of effect sizes (Supplementary Fig. 11) suggested that fetal alleles predisposing to higher PW tended to associate with higher odds of preeclampsia and a shorter gestational duration. To formally test the effect of fetal PW raising alleles on preeclampsia and gestational duration, we performed Mendelian randomization (MR) analyses. These analyses showed that fetal genetic predisposition to a higher PW raises the risk of preeclampsia (OR 1.72 [95% CI: 1.19 - 2.47] per 1 SD higher fetal genetically predicted PW, P = 6 × 10−3), and shortens gestational duration (1.9 days [95% CI: 0.74 - 3.12] shorter per 1 SD higher fetal genetically predicted PW, P = 3 × 10−3) (Supplementary Table 17, Supplementary Fig. 12). We were unable to test causal relationships between fetal genetically predicted PW and nausea and vomiting of pregnancy or IgA, because only maternal effect estimates were available for the nausea and vomiting of pregnancy outcome33 and only unadjusted fetal effect estimates for IgA37. The gestational duration results were similar to those previously seen for BW38, where fetal genetic predisposition to higher BW is associated with shorter gestation, implying a general effect of fetoplacental growth39. However, known BW-associated SNPs15 with fetal only effects showed little evidence of association in the fetal GWAS of preeclampsia (Supplementary Fig. 9t), contrary to PW-associated SNPs with fetal only effects (Supplementary Fig. 9u). MR analysis did not support a causal relationship between fetal genetic predisposition to higher BW and odds of preeclampsia (P = 0.6; Supplementary Table 17, Supplementary Fig. 12), suggesting that the increase in preeclampsia risk is more specific to placental growth.
Maternal and fetal characteristics and their effects on PW
We selected key maternal and fetal traits known to influence BW15,16 and used genetic instruments to assess whether there were similar causal relationships with PW.
Maternal glucose, fetal insulin and PW
Fetal insulin is a major determinant of fetal growth. Lower fetal insulin secretion due to rare fetal GCK mutations lead to reduced BW and PW compared with siblings without mutations40,41. To investigate the role of fetal insulin on PW in the general population, we used three SNP-sets in MR analyses: (i) 33 fasting glucose SNPs (maternal glucose crosses the placenta stimulating fetal insulin secretion; (ii) 18 insulin disposition index SNPs (estimated by insulin secretion multiplied by insulin sensitivity commonly used as a proxy for ß-cell function); and (iii) 53 BMI-adjusted fasting insulin SNPs (insulin resistance proxy in adults) (Supplementary Table 18)15. We used WLM estimates to ensure that maternal effects were adjusted for fetal effects, and vice versa. A genetic instrument representing 1 SD (0.4 mmol/l) higher maternal fasting glucose level was associated with a 47.4-g (95% CI: 23.7 - 71.2 g) higher placental weight (P = 4.46 × 10−4; Fig. 6; Supplementary Table 18). In addition, a 1-SD genetically higher fetal disposition index was associated with a 13.9-g (1.2 - 26.6) higher PW. Alleles that raise insulin secretion tend to lower glucose levels, and this was reflected in the opposite direction of the effect estimates for maternal disposition index (Fig. 6; Supplementary Table 18). The causal effect estimate for fetal insulin resistance was directionally consistent with causing lower PW, but the 95% CI crossed the null. These analyses support a role for insulin produced by the fetus, either directly or indirectly, in regulating growth of the placenta, providing a key link between fetal insulin and placental growth.

Results of two-sample MR analyses estimating the causal effects of maternal height, glycemic traits and blood pressure on PW, and the effects of fetal genetic predisposition to the same traits on PW. Maternal WLMs are adjusted for effects of the fetal genotypes, and fetal WLM analyses are adjusted for the effects of the maternal genotypes. Abbreviations: DBP, diastolic blood pressure; meta, meta analysis; SBP, systolic blood pressure; WLM, weighted linear model. Units for exposures are: height (SD), fasting glucose (SD), disposition index (SD), fasting insulin (SD), SBP (10 mmHg), DBP (10 mmHg). Causal estimates are g of placental weight per unit exposure.
Height and PW
Our analyses showed that both fetal and maternal genetic predisposition to a greater adult height were associated with greater PW, consistent with previous associations with higher BW and length15,16. A 1-SD (approx. 6 cm) higher genetically predicted adult height in the fetus was associated with a 12.1-g (95% CI: 7.1 - 17.2) higher PW (P = 2.88 × 10−6; Fig. 6; Supplementary Table 18) and a 1-SD higher maternal height was associated with a 4.7-g (95% CI: 0.8 - 8.7) higher PW, independent of direct fetal effects (P = 1.89 × 10−2; Fig. 6; Supplementary Table 18). These findings further emphasize key contributions of common fetal genetic factors to the close relationship between fetal and placental growth.
Blood pressure and PW
Higher maternal blood pressure in pregnancy is associated with reduced fetal growth42. To estimate the causal effect of maternal SBP or DBP on PW (adjusting for any direct fetal genetic effects), we used 68 known systolic blood pressure (SBP)-associated variants and 71 known diastolic blood pressure (DBP)-associated variants in two-sample MR analyses. We found no evidence of a causal effect of either maternal SBP (P = 0.23) or maternal DBP (P = 0.91; Fig. 6; Supplementary Table 18). However, analyses using direct fetal genotype effects (adjusted for maternal genotype effects) suggested that a 10-mmHg genetically higher DBP in the fetus caused a 19.0-g (95% CI: 3.8 - 34.3 g) lower PW, with a weaker effect of SBP in the same direction (9.5 g [95% CI: 0.5 - 19.7 g]; Fig. 6; Supplementary Table 18). Previous MR analyses of fetal blood pressure effects on BW have been inconsistent: those that used similar methods15,43 to the current analyses found no fetal effects, while others using transmitted and non-transmitted alleles16,38 supported fetal genetic predisposition to higher SBP being causally related to lower BW. Our findings for PW are similar to the latter.
PW and later neuropsychiatric traits
The role of placenta in neurodevelopment and risk of later psychiatric disease in the offspring is a growing research field. Using data from the Initiative for Integrative Psychiatric Research (iPSYCH) cohort (n = 100,094), we analyzed observed fetal polygenic scores (PGS) for PW in groups of individuals who later developed neuropsychiatric disease. As expected, higher deciles in the PGS distribution corresponded to higher observed PW in a population representative sample, and we also observed similar patterns of association with PW in groups of individuals with autism spectrum disorder (ASD), attention deficit/hyperactivity disorder (ADHD), affective disorders, and schizophrenia (Supplementary Fig. 13). Logistic regression of each disease on fetal PGS for PW did not show any association, although there was a nominally significant association between higher observed PW and reduced risk of ADHD (P = 0.0247; Supplementary Fig. 14).
Discussion
In this first GWAS of PW, we identify 40 independent association signals. These partially overlap with known BW loci, but 12 PW signals are related predominantly or only to PW, with connections to placental development and function. We observe a maternal POE near KCNQ1. Moreover, we find that fetal genetically mediated higher PW raises preeclampsia risk and shortens gestational duration, and we demonstrate a role for fetal insulin in regulating placental growth. Fig. 7 summarizes central results of the study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A salient feature of this study was the ability to address mode of association questions through analyses of fetal and parental genomes, application of WLM approaches, and analyses of almost 20,000 child-mother-father trios from the MoBa study. There was a clear predominance of fetal effect signals among the genome-wide significant loci, and fetal SNP heritability was almost double that of maternal SNP heritability (Supplementary Fig. 5). A total of 26 signals were classified as fetal only, and a further 3 as fetal and maternal. However, there were 4 loci that showed no fetal effect, and instead represent indirect maternal genetic effects acting on PW via the intrauterine environment. For loci also known to be associated with BW, our mode of association results were in good agreement with a previous classification16.
Investigations using MoBa trio data found a pronounced POE signal near KCNQ1 carried by the maternally transmitted allele, in agreement with previously observed silencing of the paternal alleles of KCNQ1 and the nearby CDKN1C gene. Notably, based on WLM analysis, we had classified the KCNQ1 locus as having maternal and fetal effects in the same direction. Although the WLM may improve power to distinguish between maternal and fetal effects in available samples, family studies are still key to attaining clarity about how associations with neonatal phenotypes may arise, through fetal effects, POEs, maternal effects, or a combination. The KCNQ1 finding has implications in fetal and placental growth through at least two pathways44. Placenta cells have a unique epigenetic profile that regulates its transcription patterns that may be associated with adverse pregnancy outcomes if disturbed. Beckwith-Wiedemann syndrome is an overgrowth syndrome with elevated PW and BW44. Approximately 50% of these patients have lost maternal-specific methylation at the putative Imprinting Control Region 2 (ICR2) within intron 10 of KCNQ1 that is also the transcriptional start site of the regulatory long non-coding RNA KCNQ1OT145 controlling expression of CDKN1C and KCNQ1. The location of the lead signal, rs2237892, to this region suggests it influences PW through an effect on methylation at ICR2 in the placenta. Known associations between SNP rs2237892 and altered insulin secretion46,47 support a role for fetal insulin in mediating the association with PW. Additionally, placental DNA methylation at KCNQ1 has been associated with maternal insulin sensitivity48, influencing maternal glucose availability across the placenta, and stimulating fetal insulin thus leading to fetal and placental growth.
Our results support a role for fetal genetic factors that predispose to a higher PW in raising preeclampsia risk: for every 1-SD higher genetically mediated PW, there was a more than 1.5-fold higher odds of preeclampsia. Preeclampsia is a complication that occurs in 3-5% of pregnancies4,49, but is poorly understood. The causal relationship we observed appears specific to placental growth as there was no similar effect of fetal genetic predisposition to higher BW. This finding is initially counter-intuitive, considering that preeclampsia is often linked with fetal growth restriction. However, the relationship between preeclampsia and placental weight is not consistent: early-onset preeclampsia, which is likely caused by defective placentation, is frequently associated with fetal growth restriction and low placental weight50,51, while the more common, “late-onset” or “term” preeclampsia (delivery in or after gestational week 37) has been associated observationally with both lower and higher PW51. Late-onset preeclampsia is thought to result largely from interactions between aging of the placenta and a maternal genetic susceptibility to cardiometabolic diseases50, but there is evidence that fetal trophoblast function is also important: the common genetic contributor to preeclampsia at the FLT1 locus (lead SNP rs4769613) is fetal. Fetal FLT1 likely mediates maternal endothelial dysfunction, but the variant was not associated with PW in our study, or with BW (P > 0.05)15. Our finding is supportive of a role for increased fetoplacental demands, which may result in uteroplacental mismatch, leading to late-onset preeclampsia4 and supports the role of fetally-mediated placental mechanisms in contributing to this condition.
Using PW as a proxy for placental growth had the advantage to reach the large sample size of this study, leveraging routine records in birth registers, and thereby yielding sufficient statistical power for discovery. One limitation, however, is that PW measured after delivery only crudely proxies placental growth and does not directly capture placental insults or other indications of placental dysfunction. Our study would therefore be complemented well by other approaches, e.g., involving single-cell transcriptomics of placental cells early in pregnancy19, characterization of placental mosaicism52, sequencing of cell-free RNA transcripts of placental origin in maternal circulation during pregnancy53, and RNA sequencing of placental samples without and with pregnancy complications25,54. Furthermore, while our results point to plausible candidate genes (e.g., FCGR2A, FCGR2B, TBX20, SERPINA1, SLC7A5) and potential effects on placental development and morphology, and transport of antibodies and amino acids, follow-up studies are required to establish causal links and to illuminate mechanisms related to placental growth.
Our study complements large-scale GWAS of BW. While genetic correlation and colocalization analyses revealed an overlap between signals associated with PW and BW, there were also notable differences. Twelve loci were classified as only or predominantly PW, and one signal showed opposite directions of effects between PW and BW, emphasizing the complexity of common and distinct genetic regulation and physiological processes affecting placental and fetal growth. Thus, our findings provide an improved understanding of the role of placenta for fetal growth and biological processes and complications such as gestational duration and preeclampsia. Future research may focus on additional proxies for placental function and environmental influences, as well as the role of placenta for later-life health outcomes.
Methods
Study cohorts and phenotype handling
Studies of individuals from European populations conducted genome-wide association analysis (GWAS) of placental weight as part of the EGG (Early Growth Genetics) Consortium. Where data was available, multiple births, congenital anomalies and babies born before 37 completed weeks of gestation or after 42 weeks and 6 days of gestation were excluded. Additionally, placental weight values < 200 g or > 1,500 g, or greater than five standard deviations from the mean were excluded. Placental weight was Z-score transformed for analysis. Details for phenotype exclusions, data collection, sample size, mean placental weight and gestational age for each cohort can be found in Supplementary Tables 1, 3, 5.
Fetal, maternal, and paternal meta-analyses of PW GWAS
We performed separate GWAS to test associations between PW and fetal, maternal, and paternal genotypes, as detailed in equations (1) to (3).
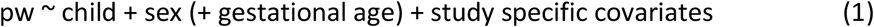
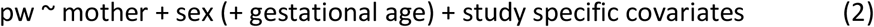
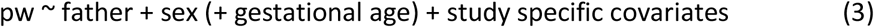 where ‘pw’ refers to the standardized PW, and ‘child’, ‘mother’ and ‘father’ refer to the number of tested alleles (as genotype probabilities) for a given variant in the child, mother, and father genomes, respectively.
where ‘pw’ refers to the standardized PW, and ‘child’, ‘mother’ and ‘father’ refer to the number of tested alleles (as genotype probabilities) for a given variant in the child, mother, and father genomes, respectively.
Analyses were conducted twice, once adjusting for sex and gestational age and once adjusting for sex only. Adjustment was also made for ancestry principal components, and the number of principal components included was determined on a per-cohort basis. Genotypes were imputed to the HRC reference panel in most studies, with exceptions noted in Supplementary Tables 2, 4, 6. Details of imputation and analysis for individual studies are shown in Supplementary Tables 2, 4, 6. Association results from fetal, maternal, and paternal GWASs using both adjustment strategies were combined separately in fixed-effects meta-analyses implemented in METAL55, resulting in six meta-analyses. Meta-analyses were performed independently by two analysts. SNPs were excluded if they were present in fewer than two studies or the number of individuals for the SNP was < 5,000. Genome-wide significant loci were defined as regions with one or more SNP with P < 5 × 10−8, and these SNPs were defined as belonging to different loci if the distance between them was > 500 kb. The lead SNP at each locus was the one with the smallest P-value. Secondary signals within each locus were identified using approximate conditional and joint multiple-SNP (COJO) analysis performed using GCTA-COJO56. Independent SNPs were defined as those with conditional P < 5 × 10−8. The linkage disequilibrium reference panel was made up of 344,241 individuals from the UK Biobank defined as having British ancestry57.
Genomic SEM
To calculate the SNP heritability of the maternal, paternal and offspring contributions we used a framework within genomic SEM20 developed by Moen et al21. In short the genomic SEM method20 involves two stages. In the first stage, LD score regression methods using pre-computed LD Scores from a European population provided by the original developers of LD score regression24,58 are applied to GWAS summary results statistics to estimate the genetic variance of each trait, and the genetic covariance between traits. In the second stage of genomic SEM, a user-defined SEM is fit to the genetic covariance matrix and parameters and their standard errors are estimated.
The genomic structural equation model we used to partition genetic covariances into maternal and offspring components is displayed in Supplementary Fig. 15. Results from the three placenta weight GWAS (squares) were modelled in terms of latent maternal, paternal and offspring genetic variables (circles). The lower part of this model reflects simple biometrical genetics principles (i.e. the fact that offspring and maternal genome are correlated 0.5) and consists of path coefficients fixed to the value one or one half. The top half of the model consists of free parameters requiring estimation - three SNP heritabilities (one for each trait), and three genetic covariances between the variables, representing commonalities in genetic action across the maternal, paternal and offspring genomes21.
It is important to realize that fitting a complicated SEM like the one in Supplementary Fig. 15 is necessary to obtain asymptotically unbiased estimates of SNP heritabilities and genetic correlations. The reason is that GWAS of perinatal traits represent a complicated mixture of maternal, paternal and offspring genetic effects. Our SEM disentangles these effects from each contributing GWAS. In contrast, the model underlying LD Score regression makes no allowance for this complication, and so naïve use will lead to biased estimates of SNP heritability and genetic correlations containing an unknown mixture of maternal, paternal and fetal effects21. Summary results statistics files from the GWASs described above were combined using genomic SEM20. The software was set to not exclude INDELs.
Partitioning fetal, maternal, and paternal effects, and allele transmission analysis
Partitioning fetal, maternal, and paternal effects was performed from the summary statistics obtained from the GWAS (equations (1) to (3)) using a weighted linear model (WLM) similar to that applied in Warrington et al., extended to include paternal data and overlap in individuals between GWASs. The estimates of partitioned fetal, maternal, and paternal effects ( , and
, and  respectively), estimated from the GWAS estimates (
respectively), estimated from the GWAS estimates ( , and
, and 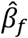 respectively) are
respectively) are
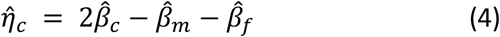
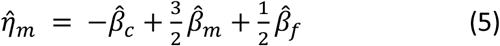
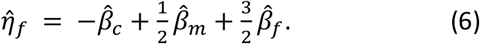
To account for sample overlap between GWASs, the covariance in estimates of the regression coefficients has the form
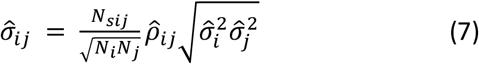 where Nsij is the number of individuals contributing to both analyses, Ni is the number of individuals contributing to analysis i,
where Nsij is the number of individuals contributing to both analyses, Ni is the number of individuals contributing to analysis i,  is the correlation between the estimates
is the correlation between the estimates  and
and  in the overlapping samples, and
in the overlapping samples, and  is the standard error of
is the standard error of  . The term
. The term  can be estimated as the intercept of a bivariate LDScore regression24. Standard errors for the partitioned effects are then estimated as
can be estimated as the intercept of a bivariate LDScore regression24. Standard errors for the partitioned effects are then estimated as
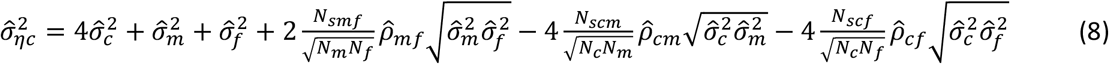
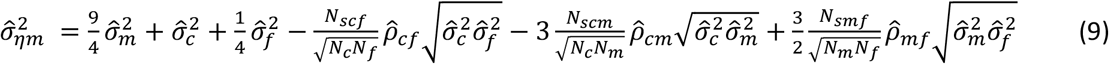
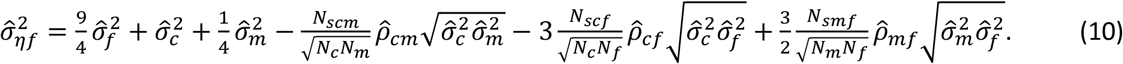
P-values are calculated from WLM estimates using a Z-test, with test statistic

To complement this analysis, we utilized the independent child-mother-father trios available in MoBa to perform conditional analyses where the association of fetal and parental genotypes with PW are conditioned against each other, as detailed in equation (12).
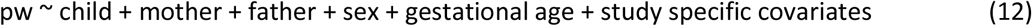
In this set of independent trios, using the phasing of the children’s genotypes, we inferred the parent-of-origin of the genotyped alleles as done by Chen et al.38. We then studied the association of the PW with maternal and paternal transmitted and non-transmitted alleles, as detailed in equation (13).
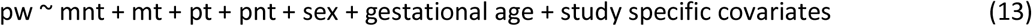
Where ‘mnt’ and ‘mt’ refer to the maternal non-transmitted and transmitted alleles, respectively; and similarly, ‘pt’ and ‘pnt’ refer to the paternal transmitted and non-transmitted alleles, respectively.
To estimate the significance of effects mediated by the maternal transmitted only, and hence test for parent-of-origin effects, we conditioned it on the genotypes of the child, mother, and father for the same variant, as detailed in equation (14).
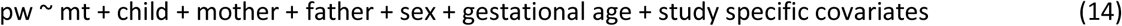
Where variables are coded as in equations (13) and (14). Note that trio and haplotype analyses in MoBa were conducted on hard-called genotypes. Sex, gestational age, and intercept were included in the model as well as study specific covariates, i.e., ten principal components and genotyping batches. The share of Mendelian errors was estimated using trios presenting at least a homozygous parent. If a variant presented more than 50 % Mendelian errors, children alleles were swapped. All estimates are provided in Supplementary Table 11.
To classify SNPs as either fetal, maternal or paternal, we used similar criteria to those used previously for BW SNPs15. We classified SNPs into three categories: (1) Fetal only: the P value for the fetal estimate is lower than a Bonferroni corrected threshold (0.05/37 = 0.00132), and the 95 % CI surrounding the estimate does not overlap the 95 % CI for the maternal estimate; (2) Maternal only: the P value for the maternal estimate is lower than a Bonferroni corrected threshold (0.00132), and the 95 % CI surrounding the estimate does not overlap the 95 % CI for the fetal estimate; and (3) Maternal and Fetal: the P value for both the maternal and fetal association estimates were less than the Bonferroni corrected threshold (0.00132). If a SNP did not fit any of these criteria it was marked as “unclassified”.
Assessing colocalization of fetal, maternal or paternal association signals
To further supplement the aforementioned classifications, we performed colocalization analysis to determine overlap between GWAS signals from fetal, maternal, and paternal meta-analyses using the R library ‘coloc’59 with the default prior probability for colocalization. Signals were defined as colocalising if the posterior probability of shared association signals (P4) was > 0.8, distinct if the posterior probability of independent signals (P3) was > 0.8, and undetermined if neither P3 or P4 was > 0.8.
LD score regression
To estimate the genetic correlation between PW and BW, linkage disequilibrium score regression24,58 was performed using summary statistics from gestational age and fetal sex adjusted analyses and BW summary statistics taken from Warrington et al15. Estimates for the results from the meta-analyses and WLM for both PW and BW were calculated for each of the fetal, maternal and paternal genomes.
Testing whether lead PW SNPs were also associated with BW
Colocalization analysis was again utilized to determine whether PW loci close to those previously identified in GWAS ofBW represent the same association signal, using the same colocalization methods as above.
To classify SNPs as placental weight or BW, we calculated the 95 % CI in SD units for the PW lead SNP for both PW and BW. We then compared the 95% CIs; if these 95% CIs did not overlap the SNP was classified as “predominantly or only PW” unless either BW association (fetal or maternal) was in the opposite direction to the PW association and its associated P value was < 0.05, in which case it was classified as PW and BW in opposite directions. SNPs whose 95% CIs for PW and BW overlapped were classified as “PW and BW same direction”.
Enrichment analysis using tissue-specific mRNA expression and scRNA data
We tested enrichment of expression of the PW associated genes in specific tissues or cell-types by comparing the ranks of gene expression across different tissue or cell-types (Supplementary Fig. 16). This method involved two steps: first, the tissue or cell-type specific expression levels from a reference expression data set were rank normalized across all tissues or cell-types for each gene; second, for a particular tissue or cell-type, expression enrichment of the test genes were compared against all other genes by the Wilcoxon rank-sum test. We used 31 protein-coding genes close to the index SNPs of the 32 PW-associated loci identified in the fetal GWAS (First section of Table 1; the LOC339593 locus was not included because no protein-coding gene nearby). For the enrichment analysis in specific tissues, we used tissue-specific mRNA expression data from the Human Protein Atlas (RNA consensus tissue gene data)29. For the enrichment analysis in different cell-types, we used the scRNA-seq data of about 70,000 single cells at the decidual-placental interface in early pregnancy19. Cell-type specific gene expression of each gene was evaluated by percentage of cells with detectable scRNA reads. Significance levels for this enrichment analysis were Bonferroni corrected by number of tissues (n = 61) or cell-types (n = 32).
Identification of placental meQTL
Using the 41 SNPs identified in the discovery meta-analyses GWASes of PW, we conducted placental methylation QTL (methQTL) analysis in 395 participants with European ancestry from the Canadian cohort of the study ‘Genetics of Glucose Regulation in Gestation and Growth’ (Gen3G)26,27. We measured DNA methylation using Illumina EPIC arrays from samples collected in the fetal-facing side of placenta. We conducted DNA methylation data processing, normalization, and cell type composition estimation as previously described26. Whole genome sequencing data were processed with the DNA-Seq v3.1.4 pipeline from GenPipes60 based on BWA_mem and GATK best practices to identify high quality SNPs on GRCh37 through joint genotyping over all samples. SNPs with a call rate above 20%, Hardy-Weinberg equilibrium p-value above 1e-6 and minor allele counts above 10 were included.
Methylation QTL were identified using beta values of 681 795 CpGs with TensorQTL61 in a cis-window of 0.5 Mb and the following covariates: four genotype PCs, fetal sex and cell type composition (estimates based on DNA methylation). In order to correct for the multiple CpGs tested genome-wide, a FDR threshold of ≤ 0.05 was applied on the q-values measured from the p-values extrapolated from the beta distribution by TensorQTL.
Mapping to pathways and traits
Variants classified as PW through the SNP classification of PW or BW, were investigated further to identify relevant pathways and related biology. Systematic look-ups using Open Targets Platform, The Human Protein Atlas, and International Mouse Phenotyping Consortium. Each variant was assessed for which gene it is functionally implicated in through information gained regarding the variant’s involvement in molecular phenotype experiments, chromatin interaction experiments, in silico function predictions and distance between the variant and the gene’s canonical transcription start site. The most likely candidate gene was then used to investigate the placenta relevant cells that the gene is expressed in using information on the Open Targets Platform and The Human Protein Atlas websites. The international Mouse Phenotyping Consortium website was used to identify mouse models that found associations with placental pathology after removal of the gene in single-gene knockout mice. Finally, further information was garnered through searching publications for which the candidate gene was specifically implicated in placental biology.
Lookups of SNPs in GWAS of other phenotypes
We looked at associations between our independent PW-associated SNPs and other phenotypes within the phenoscanner62. We looked for associations with the SNP itself, SNPs in LD (r2 = 1, and r2 ⋝ 0.8). We additionally looked at associations between the independent PW SNPs in GWAS for nausea and vomiting of pregnancy, hyperemesis gravidarum33, preeclampsia34, gestational duration35, miscarriage (recurrent and spontaneous)36, and 10 cytokines assayed from neonatal blood spots37 (Supplementary Table 17).
Mendelian randomization analysis
We then performed two-sample Mendelian randomization analyses with own and offspring PW as outcomes. The exposures used included height, fasting glucose, disposition index of insulin secretion, insulin sensitivity, systolic blood pressure (SBP) and diastolic blood pressure (DBP) using the same genetic instruments as the previous GWAS of BW (Supplementary Table 18)15. We estimated both the effect of each maternal exposure on PW, and also the effect of the fetal genetic predisposition to each exposure on PW. For the latter, we made the assumption that SNP-exposure effects in the fetus would be the same as in the adult samples in which they were identified. Effect sizes were converted to g using the value of 1 SD of PW of 132.5 g. We additionally used two-sample Mendelian randomization with its own PW as the exposure, and gestational age, and preeclampsia as outcomes. The SNP-PW associations were taken from our GWAS of PW, and the WLM-adjusted PW analysis. The SNP associations for all other traits were taken from external sources (Supplementary Table 17). To estimate the independent fetal associations for gestational age and preeclampsia, we first transformed the preeclampsia log odds ratios to the liability scale63, then applied the WLM in the same way as for PW. The preeclampsia independent fetal effects were then transformed to log odds ratios for analysis. A population prevalence for preeclampsia of 4.6% was used for the transformations64. We applied the inverse-variance weighted Mendelian randomization method, with MR-Egger65, Weighted Median, and Penalised Weighted Median66 acting as sensitivity analysis to test for robustness to Mendelian randomization assumptions.
Polygenic scores
To investigate a possible association between PW and future risk of neuropsychiatric disease, we conducted analyses in the Initiative for Integrative Psychiatric Research (iPSYCH) cohort. The iPSYCH cohort is a Danish population based case–cohort sample including 141,215 individuals, out of which 50,615 constitute a population-representative sample and the remainder are individuals with one or more neuropsychiatric disease diagnoses67. After restriction to individuals of European ancestries born at gestational ages between 37 and 42 weeks, 100,094 individuals remained for analysis, including 20,328 cases of ADHD, 28,672 cases of affective disorder, 17,362 cases of autism spectrum disorder, 4,052 cases of schizophrenia, and population-based sample of 32,995 individuals without any one of the four disorders. Placental weight data from the Danish Medical Birth Register were available for 33,035 of these individuals (9,200 cases of ADHD, 2,946 cases of affective disorder, 9,678 cases of autism spectrum disorder, 239 cases of schizophrenia, and 13,113 controls).
We redid the fetal meta-analysis for PW excluding the iPSYCH data, and based on the resulting summary statistics and a genetic correlation matrix created from the iPSYCH genotypes, we used LD-pred2-auto68 to generate polygenic scores (PGS) for PW. Next, we regressed observed PW on ten fetal PW PGS quantiles, including sex, gestational age and ten principal components as covariates in the model. The linear regressions were done separately for the population controls and the four neuropsychiatric diseases. We also used logistic regression to assess the association between either 1) observed PW, or 2) fetal PGS of PW, and future risk of neuropsychiatric disease, including sex, gestational age and ten principal components as covariates in the model. In these analyses, the observed PW and the fetal PGS were first standardized to have mean 0 and variance 1.
Data Availability
Individual cohorts contributing to the meta-analysis should be contacted directly as each cohort has different data access policies. GWAS summary statistics from this study are available via the EGG website (https://egg-consortium.org/).
URLs
https://platform.opentargets.org/
https://www.mousephenotype.org/
Data availability
Individual cohorts contributing to the meta-analysis should be contacted directly as each cohort has different data access policies. GWAS summary statistics from this study are available via the EGG website (https://egg-consortium.org/).
Ethics
Details of ethical approvals for the contributing studies are found in the Supplementary Information.
Author contributions
The central analysis and manuscript drafting team included R.N.B., C.F., M.Vaudel, X.W., L.S., G.-H.M., D.M.E., B.J., M.-R.J., G.Z., M.-F.H, S.J., R.M.F., B.F., P.R.N.
Sample collection, genotyping and/or phenotyping were performed and/or directed by C.F., M.Vaudel, G.- H.M., Ø.H., P.S.-N., K.B., D.W., J.R.B., H.C., K.Hao, O.A.A., B.O.Å., M.A., L.Bhatta, L.Bouchard, B.M.B, S.B., J.B.-G., P.E., L.E., C.Erikstrup, M.E., S.F., R.G., F.G.E., J.G., D.M.H., E.K., C.S.M., E.A.N., M.N., C.N.A.P., O.B.P., F.R., B.M.S., C.S., I.S., L.W.T., H.U., M.Vaarasmaki, B.J.V., C.J.W., T.A.L., D.J.G.-B., M.B., T.H., E.R.P., R.R., S.R.O., C.E.P., V.W.V.J., J.F.F., A.T.H., M.M., D.A.L., K.Hveem, T.W., H.S.N., P.M., B.J., M.-R.J., M.-F.H., S.J., R.M.F., B.F., P.R.N.
Statistical analysis was performed by R.N.B., C.F., M.Vaudel, X.W., J.C., G.-H.M., L.S., C.Albiñana, J.R., J.F., S.E.S., K.T., C.A.W., S.Srinivasan, C.S.S., J.R.B., C.Allard, M.G., T.K., D.J.L., F.W., H.C., K.Hao, M.A., F.G.E., J.G., B.J.V., T.A.L., M.B., J.F.F., D.M.E., G.Z., B.F.
Data was interpreted by R.N.B., C.F., M.Vaudel, X.W., L.S., G.-H.M., J.C., Ø.H., P.S.-N., C.Albiñana, J.R., K.T., S.Srinivasan, C.Allard, M.G., F.W., P.-É.J., B.O.Å., L.Bhatta, B.M.B., C.Ebbing, P.E., S.F., J.G., E.K., C.N.A.P., F.R., S.Sebert, M.Vaarasmaki, C.J.W., E.R.P., R.R., V.W.V.J., J.F.F., D.A.L., K.Hveem, D.M.E., B.J., M.-R.J., G.Z., M.-F.H., S.J., R.M.F., B.F., P.R.N.
All authors contributed to the final version of the manuscript.
Competing interests
O.A.A. is a consultant to HealthLytix. B.J.V. is on Allelica’s scientific advisory board. C.J.W.’s spouse works for Regeneron Pharmaceuticals. D.A.L. has received support from Roche Diagnostics and Medtronic Ltd for research unrelated to that presented here. H.S.N. has received speaker’s fees from Ferring Pharmaceuticals, Merck A/S, Astra Zeneca, Cook Medical, Ibsa Nordic.
Acknowledgements
We are grateful to Drs. Mirjana Efremova and Sarah A. Teichmann who shared the maternal-fetal interface single cell RNA data. Full acknowledgements and details of supporting grants are found in the Supplementary Information.
Footnotes
↵65 A list of members and affiliations appears in the Supplementary Note
** These authors jointly directed this work
References



