
Abstract
Background Autism spectrum disorder (ASD) is a heterogenous multifactorial neurodevelopmental condition with a significant genetic susceptibility component. Thus, identifying genetic variations associated with ASD is a complex task. Whole-exome sequencing (WES) is an effective approach for detecting extremely rare protein-coding single-nucleotide variants (SNVs). However, interpreting the functional and clinical consequences of these variants requires integrating multifaceted genomic information.
Methods We compared the effectiveness of three bioinformatics tools in detecting ASD candidate SNVs from WES data of 250 ASD family trios registered in the National Autism Database of Israel. We studied only rare (<1% population frequency), proband-specific SNVs. The pathogenicity of SNVs, according to the American College of Medical Genetics (ACMG) guidelines, was evaluated by the InterVar and TAPES tools. In addition, likely gene-disrupting (LGD) SNVs were detected based on an in-house bioinformatics tool, designated Psi-Variant, that integrates results from seven in-silico prediction tools. We compared the effectiveness of these three approaches – and their combinations – in detecting SNVs in high-confidence ASD genes.
Results Overall, 605 SNVs in 499 genes distributed in 193 probands were detected by these tools. The overlap between the tools was 64.1%, 17.0%, and 21.6% for InterVar–TAPES, InterVar–Psi-Variant, and TAPES–Psi-Variant, respectively. The intersection between InterVar and Psi-Variant (I⋂P) was the most effective approach in detecting ASD genes (OR = 5.38, 95% C.I. = 3.25–8.53). This combination detected 102 SNVs in 99 genes among 80 probands (approximate 36% diagnostic yield).
Conclusions Our results suggest that integration of different variant interpretation approaches in detecting ASD candidate SNVs from WES data is superior to each approach alone. Inclusion of additional criteria could further improve the detection of ASD candidate variants.
Background
Autism spectrum disorder (ASD) comprises a collection of heterogeneous neurodevelopmental disorders that share two behavioral characteristics—difficulties in social communication, and restricted, repetitive behaviors and interests [1, 2]. The etiology of ASD has a significant genetic component, as is evident from multiple twin and family studies [3–6]. Yet, over the years, very few genetic causes of ASD have been discovered; thus, today, despite extensive research, an understanding of the overall genetic architecture of ASD remains obscure [6, 7].
The emergence of next-generation sequencing (NGS) approaches in the past decade has transformed the genetic research of complex traits [8]. These NGS technologies have facilitated high-throughput DNA sequencing for large cohorts of patients, allowing the comparison of multiple single-nucleotide variants (SNVs) between large groups of patients [9–12]. In this realm, whole-exome sequencing (WES) is particularly suitable for studying the genetics of heterogenous traits such as ASD, as it focuses on a relatively limited number of protein-coding SNVs [9–11,13–17].
Understanding the functional consequences of coding SNVs is, however, not a trivial task. In 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published standards and guidelines to generalize sequence variant interpretation and to address the issue of inconsistent interpretation across laboratories [8]. The resulting system for classifying SNVs recommends 28 criteria (16 for pathogenic and 12 for benign SNVs) and provides a set of scoring rules based on variant population allele frequency, variant functional annotation, variant familial segregation, etc. [8,18]; SNVs are classified as pathogenic (P), likely pathogenic (LP), variants of uncertain significance (VUS), likely benign (LB) or benign (B). Subsequently, multiple in-silico tools were developed to implement these ACMG/AMP criteria for annotating the prospective pathogenicity of SNVs detected in WES studies.
While the ACMG/AMP scoring approach is highly effective for detecting de-novo highly penetrant mutations for rare Mendelian disorders, it is less suitable for detecting inherited partially penetrant SNVs [19]. Such SNVs, which are usually annotated as VUS in terms of the ACMP/AMP criteria, are expected to contribute significantly to the risk of developing neurodevelopmental conditions, including ASD [9,16,17,20,21]. Thus, relying solely on the ACMG/AMP criteria for variant annotation in WES studies of ASD may result in under-representation of susceptibility SNVs, which will lead to a lower diagnostic yield for ASD. To overcome this potential limitation, we have developed “Psi-Variant,” as a pipeline to detect different types of likely gene-disrupting (LGD) SNVs, including protein truncating and deleterious missense SNVs. We applied Psi-Variant – in comparison with InterVar and TAPES, two SNV interpretation tools that use the ACMG/AMP criteria – to a large WES dataset of ASD to evaluate the concordance between these tools to detect SNVs and to assess their effectiveness in detecting ASD susceptibility SNVs.
Methods
Study sample
The study sample comprised 250 children diagnosed with ASD who are registered in the National Autism Database of Israel (NADI) [22,23] and whose parents gave consent for participation in this study. Based on our clinical records, none of the parents in the study has been diagnosed with ASD, intellectual disability, or any other type of neurodevelopmental disorder. Genomic DNA was extracted from saliva samples that were collected from participating children and their parents using Oragene®•DNA (OG-500/575) collection kits (DNA Genotek, Canada).
Whole exome sequencing
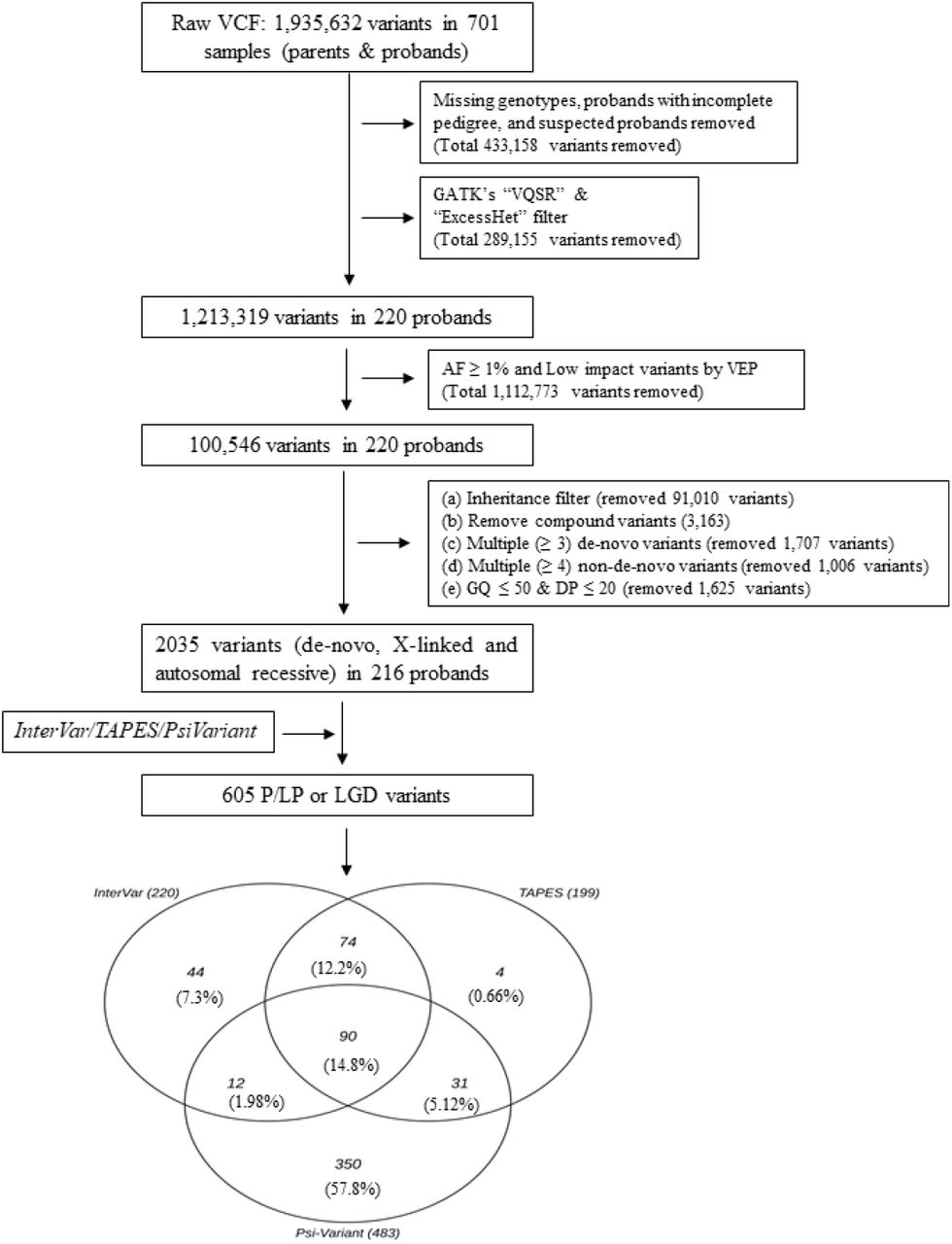
WES analysis was performed on the above-mentioned samples with Illumina HiSeq sequencers, followed by the Illumina Nextera exome capture kit, at the Broad Institute as part of the Autism Sequencing Consortium, as described previously [11]. Sequencing reads aligned to Genome Reference Consortium Human Build 38 and aggregated into BAM/CRAM files were analyzed using the Genome Analysis Toolkit (GATK) [24] to generate a joint variant calling format (vcf) file for all subjects in the study. We excluded data for 30 probands from the raw vcf file due to incomplete pedigree information or to low-quality WES data. Thus, WES data for 220 ASD trios were analyzed in this study (Fig. 1).

Analysis workflow for detecting LP/P/LGD SNVs from the WES data. LP/P SNVs were detected by InterVar and TAPES by implementing ACMG/AMP criteria. LGD SNVs were detected by Psi-Variant by utilizing in-house criteria.
Data analysis
The SNV detection process in this study is outlined in Fig. 1. and explained below.
Data cleaning
The raw vcf file contained 1,935,632 SNVs. From this file, we removed SNVs with missing genotypes and/or SNVs in regions with low read coverage (≤ 20 reads), and/or with low genotype quality (GQ ≤ 50). In addition, we removed all common SNVs (i.e., those with a population minor allele frequency >1%) [25] as well as those that did not pass the GATK’s “VQSR” and “ExcessHet” filters. Thereafter, we used an in-house machine learning (ML) algorithm to remove other potentially false-positive SNVs. The details of this ML algorithm and its efficiency in classifying between true positive and false positive SNVs are summarized in the supplementary file. Finally, we used the pedigree structure of the families to identify proband-specific genotypes, including de-novo SNVs, recessively inherited SNVs, and X-linked SNVs (in males). From these genotypes, we removed multiallelic SNVs and those that were classified as “de-novo” and appeared in more than two individuals in the sample.
Identifying ASD candidate SNVs
We searched for candidate ASD SNVs using three complementary approaches. First, we applied InterVar [18] and TAPES [26], two commonly used publicly available command-line tools that use ACMG/AMP criteria [8], to detect LP/P SNVs. In addition, we assigned the ACMG/AMP PS2 criterion to all the de-novo SNVs to detect additional LP/P SNVs from the list of VUS. Since both InterVar and TAPES are less sensitive tools for detecting recessive possible gene disrupting (LGD) SNVs [19], we developed an integrated in-house tool, designated Psi-Variant, to detect LGD SNVs. The Psi-Variant workflow starts by using Ensembl’s Variant Effect Predictor (VEP) [25] to annotate the functional consequences for each variant in a multi-sample vcf file. Then, all frameshift indels, nonsense and splice acceptor/donor SNVs are further analyzed by the LoFtool [27] with scores of < 0.25 are annotated as intolerant SNVs. In addition, it applies six different in-silico tools to all missense substitutions and annotates them as “deleterious/damaging” if at least three (≥ 50%) of them were exceeded the following cutoffs: SIFT [28] (< 0.05), PolyPhen-2 [29] (≥ 0.15), CADD [30] (> 20), REVEL [31] (> 0.50), M_CAP [32] (> 0.025) and MPC [33] (≥ 2). These scores were extracted by utilizing the dbNSFP database [34].
Comparison between InterVar, TAPES and Psi-Variant
We compared the number of SNVs detected by each of the three tools as well as percentages of SNVs that were detected by different combinations of these tools. Thereafter, we used the list of ASD genes (n = 1031) from the SFARI Gene database [35] (accessed on 11 January 2022) as the gold standard to test the effectiveness of these combinations. We computed the odds ratio (OR) and positive predictive value (PPV) for detecting candidate ASD SNVs in SFARI genes.
Software
Data storage, management, and analysis were conducted on a high-performing computer cluster in a Linux environment using Python version 3.5 and R Studio version 1.1.456. All the statistical analyses and data visualizations were incorporated in R Studio.
Results
Detection of candidate SNVs by the different tools
A total of 605 SNVs in 193 probands were detected by InterVar (n = 220), TAPES (n = 199) or Psi-Variant (n = 483) from a dataset of 2,035 high-quality, ultra-rare SNVs with proband-specific genotypes (Fig. 1). Of these, 90 SNVs (14.9%) were detected by all three tools. The highest concordance in detected SNVs was observed between InterVar and TAPES (64.3%), followed by TAPES and Psi-Variant (21.6%) and InterVar and Psi-Variant (17.0%).
The characteristics of the detected SNVs are shown in Table 1. Significantly higher rates of LoF and missense SNVs were detected by all three tools compared to the rates of these SNVs in the clean vcf file (P < 0.001). As expected, missense variants comprised the majority of detected SNVs, with 81.6%, 58.8% and 51.4% of the SNVs detected by Psi-Variant, TAPES, and InterVar, respectively. Notably, a higher number of frameshift SNVs were detected by Psi-Variant than by InterVar and TAPES (58 vs. 39 and 22, respectively), but the percentages of these SNVs out of the total number of detected SNVs was lower due to the markedly higher number of SNVs detected by Psi-Variant.
Characteristics of the detected SNVs by InterVar, TAPES, and Psi-Variant from the WES data
Notably, almost all (≥ 95%) SNVs detected by either InterVar or TAPES were de-novo SNVs, while de-novo SNVs comprised only 36.2% of the SNVs detected by Psi-Variant, which also detected a high portion of X-linked and autosomal recessive SNVs (37.1% and 26.7%, respectively). Examination of the distribution of the detected SNVs in genes associated with ASD according to SFARI Gene database [35] revealed a two-fold enrichment of SNVs distributed in ASD genes (for all detection tools) compared to their portion in the clean vcf file, and even a higher enrichment of SNVs distributed in high-confidence ASD genes (P < 0.001).
Effectiveness of ASD candidate SNVs detection
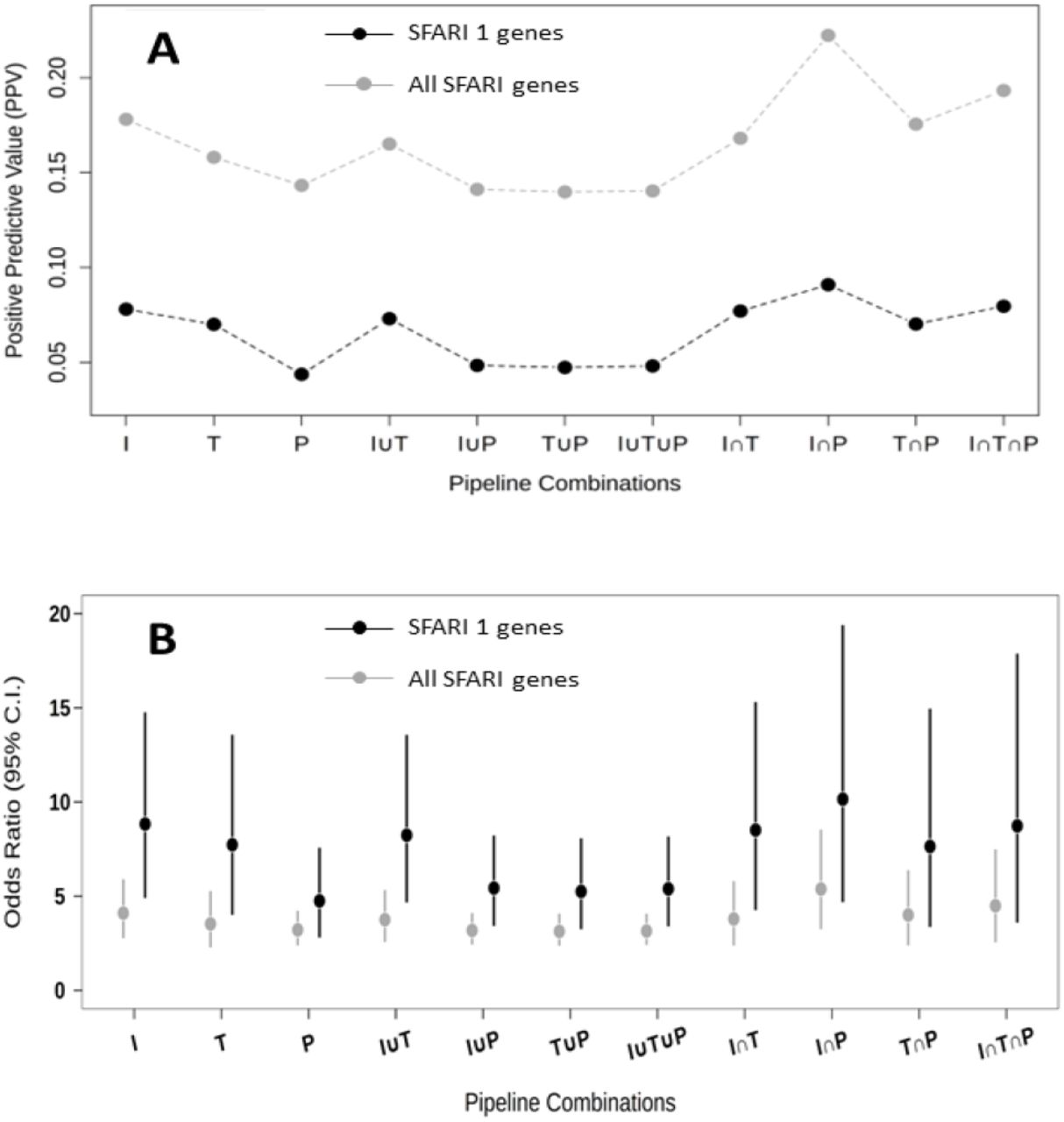
To assess the effectiveness of the different tools in detecting ASD candidate SNVs, we calculated the PPV and the OR for detecting ASD genes (i.e., those listed in the SFARI Gene database [35]) for different combinations of utilization of the three tools. The results of these analyses are shown in Fig. 2. Utilization of any of the three tools resulted in a significant enrichment of ASD genes, with the highest enrichment being observed in SNVs detected by InterVar (PPV = 0.178; OR = 4.10, 95% confidence interval (C.I.) = 2.77–5.90) followed by TAPES (PPV = 0.158; OR = 3.53, 95% C.I. = 2.28–5.27) and Psi-Variant (PPV = 0.143; OR = 3.21, 95% C.I. = 2.39–4.22). Notably, better performance in detecting ASD candidate SNVs was obtained at the intersection of the detected SNVs between InterVar and Psi-Variant (I ⋂ P) (PPV = 0.222; OR = 5.38, 95% CI = 3.25–8.53). The I ⋂ P combination was also the most effective in detecting SNVs in high-confidence ASD genes (i.e., those with score of 1 in the SFARI Gene database [35] (Fig. 2). Overall, the I ⋂ P combination detected 102 SNVs distributed in 99 genes (22 SFARI genes) in 80 probands (a detection yield of 36%).

{kind=link}
{kind=link}
Effectiveness of InterVar (I), TAPES (T), Psi-Variant (P), and their combinations in detecting candidate variants in ASD genes. A Positive predictive value (PPV) of detecting candidate variants in SFARI 1 and all SFARI genes. B Odds Ratios (ORs) of detecting candidate variants in SFARI 1 and all SFARI genes.
Discussion
In this study, we assessed the concordance and effectiveness of three bioinformatics tools in the interpretation of SNVs detected in WES of children with ASD. There was better agreement in variant detection between InterVar and TAPES than between Psi-Variant and each of these two tools, probably because both InterVar and TAPES are based on the ACMG/AMP guidelines [8], while Psi-Variant uses the interpretation of six in-silico tools in assessing the functional consequence of LGD SNVs. In addition, most (94%) of the SNVs detected by either InterVar or TAPES were de-novo SNVs, compared to only 36% of the SNVs detected by Psi-Variant. This difference may be attributed to the fact that ACMG/AMP guidelines are particularly designed to detect de-novo highly penetrant SNVs, while inherited SNVs (autosomal recessive and X-linked) are usually classified as VUS [19]. Importantly, such rare inherited SNVs have been found to be associated with a variety neurodevelopmental conditions, including ASD [9,16,17,20,21]. Another major difference between these tools lies in the detection of in-frame insertions/deletions that comprised ∼20% of the SNVs detected by either InterVar or TAPES, while such SNVs were discarded by Psi-Variant. We decided to exclude these SNVs from Psi-Variant because their clinical relevance has been demonstrated in several genetic disorders [36,37] but not in ASD [38–40].
Another important factor that could affect the concordance between the three tools is the annotation tools they use. Specifically, both InterVar and TAPES use AnnoVar [41] for their variant annotation, while Psi-Variant uses Ensembl’s VEP [25]. It has already been shown that AnnoVar and VEP have a low concordance in the classification of LoF SNVs [42]. In addition, each tool, InterVar, TAPES, and Psi-Variant, utilizes a different set of in-silico tools for the classification of missense SNVs, with SIFT [28] alone being shared by all three tools. These differences are probably the reason for the large differences in the detection of missense SNVs between the three tools (Table 1).
Today, there are no accepted guidelines for the detection of ASD susceptibility SNVs from WES data. Therefore, many genetic labs use the ACMG/AMP guidelines [8] for this purpose. Our findings suggest that combining these guidelines with other variant classification criteria improves detection of ASD susceptibility SNVs. Thus, this approach could be applied in the future for drawing up specific guidelines for the detection of ASD susceptibility SNVs.
Of note, 77.5% of the SNVs detected by the most effective integrative pipeline (I ⋂ P) affect genes with no known association to ASD, according to the SFARI Gene database [35]. This finding highlights the capability of the integrative pipeline (I ⋂ P) to detect of novel ASD genes. Obviously, the association of these genes and SNVs with ASD susceptibility needs to be validated in additional studies.
The results of this study should be considered under the following limitations. First, the effectiveness assessments of the different tools and their combinations were based on ASD genes from the SFARI Gene database [35]. While this is the most commonly used database for ASD genes and is continuously updated, it is based on data curated from the literature and may thus include genes that were falsely associated with ASD. Second, the variant detection analyses were performed on WES data of a cohort from the Israeli population, which may not necessarily be representative of the genetic architecture of ASD. Third, the tools used in this study were designed to detect only extremely rare SNVs with relatively large functional effects. Thus, a more effective approach for detection of ASD susceptibility variants should also include interpretation of copy-number variants [43–46] and other variants with milder functional effects [16,47,48] Finally, it should be noted that there are many other approaches for variant interpretation from WES data. Thus, it is possible that combinations of other approaches would be more effective in the detection of ASD susceptibility variants from WES data than the approaches investigated in this study.
Conclusions
Our findings suggest that an integration of different variant interpretation approaches is more effective in the detection of ASD candidate SNVs from WES data than each of the examined approaches alone. Inclusion of additional criteria to this integrative approach may further improve its effectiveness in the detection of ASD candidate variants.
Data Availability
WES data were generated as part of the ASC and are available in dbGaP with study accession: phs000298.v4.p3. The generated results and codes are available in a GitHub public repository: https://github.com/AppWick-hub/Psi-Variant or available upon reasonable request to the corresponding author Prof. Idan Menashe (idanmen{at}bgu.ac.il).
Declarations
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Soroka University Medical Center (SOR-076-15; 17 April 2016).
Ethics approval and consent to participate
Informed consent was obtained from all the families involved in the study.
Consent for publication
All the data from the registered families presented here are de-identified.
Availability of data and materials
WES data were generated as part of the ASC and are available in dbGaP with study accession: phs000298.v4.p3. The generated results and codes are available in a GitHub public repository: https://github.com/AppWick-hub/Psi-Variant or available upon reasonable request to the corresponding author Prof. Idan Menashe (idanmen{at}bgu.ac.il).
Competing interests
The authors declare no competing interests.
Funding
This study was supported by a grant from the Israel Science Foundation (1092/21).
Authors’ contributions
Conceptualization: A.S. and I.M.; methodology: A.S. and I.M.; software: A.S. and L.L.; validation: A.S. and I.M.; formal analysis: A.S.; resources: H.G., G.M., A.M., Y.T., A.A. and I.D.; data curation: A.S.; writing—original draft preparation: A.S. and I.M.; writing—review and editing: I.M., and A.S.; supervision: I.M.; project administration: I.M.; funding acquisition: I.M. All the authors have read and agreed to the published version of the manuscript.
Acknowledgements
We thank the families who participated in this research, without whose contributions genetic studies would be impossible.
List of abbreviations
- ACMG/AMP
- American College of Medical Genetics and Genomics/Association of Molecular Pathology
- ASD
- autism spectrum disorder
- C.I.
- confidence interval
- GATK
- Genome Analysis Toolkit
- LGD
- likely gene disrupting
- LoF
- loss of function
- LP
- likely pathogenic
- ML
- machine learning
- NADI
- National Autism Database in Israel
- NGS
- next-generation sequencing
- OR
- odds ratio
- P
- pathogenic
- PPV
- positive predictive value
- SNV
- single nucleotide variants
- VEP
- Variant Effect Predictor
- vcf
- variant calling format
- VUS
- variants of uncertain significance
- WES
- whole exome sequencing
Reference



