
ABSTRACT
Objective Identifying microbial targets in irritable bowel syndrome (IBS) is challenged by dynamic microbiota-metabolite-host interactions. We aimed to assess microbial features associated with short chain fatty acids (SCFA) and determine if features were related to IBS symptoms, subtypes, and endophenotypes.
Design We performed an observational study of stool microbial metagenomes, stool SCFA, and IBS traits (stool form, stool bile acids, and colonic transit) in patients with IBS (IBS with constipation [IBS-C] IBS with diarrhea [IBS-D]) and healthy controls. We analyzed associations of microbiome composition with stool SCFA to identify microbe-SCFA relationships that were shared and distinct across groups. We compared gut microbiome-encoded potential for substrate utilization across groups and within a subset of participants selected by stool characteristics. In IBS-D, we compared stool microbiomes of patients with and without bile acid malabsorption (BAM).
Results Overall stool microbiome composition and abundances of individual taxa differed between groups. Increased abundances of several bacterial species were observed in IBS-D including Dorea sp. CAG:317.. Microbes-SCFA relationships varied across groups after accounting for transit and bile acids. Significant microbe-SCFA were common in IBS-D and several SCFA-producing species were inversely correlated with SCFA. Among participants selected by stool form characteristics, functional profiling demonstrated differential abundances of microbial genes/pathways for SCFA metabolism and degradation of carbohydrates and mucin across groups. SCFA-producing taxa were reduced in IBS-D with BAM.
Conclusion Microbe-SCFA associations differ across IBS subtypes and traits. Altered substrate preferences offer insights into functional microbiome traits and could be used as novel microbial IBS biomarkers.
KEY MESSAGES What is already known on this topic: The intestinal microbiota and its metabolites (e.g., short chain fatty acids [SCFA]) modulate irritable bowel syndrome (IBS) pathophysiology.
What this study adds: We studied microbe-SCFA associations across IBS subtypes and endophenotypes to demonstrate (1) the intestinal microbiome plays distinct roles across IBS subtypes, (2) microbial substrate preferences vary between IBS subtypes and influences stool form, and (3) microbe-SCFA patterns may reveal key taxa that underlie shared and distinct microbial mechanisms across the IBS spectrum.
How this study might affect research, practice or policy: Findings demonstrate that structural and functional features of the intestinal microbiome may represent unbiased microbial biomarkers for clinical and mechanistic IBS subtypes. Further study of these putative microbial targets as well as their interactions with diet- and host-specific traits should be pursued to develop individualized microbiome-based approached to IBS management.
INTRODUCTION
Irritable bowel syndrome (IBS) is a burdensome disorder of gut-brain interaction with an estimated global prevalence rate of 5-10%.1 Pathophysiological mechanisms of IBS include disturbances in motility or transit, altered intestinal secretion, impaired intestinal permeability, immune cell reactivity, visceral hypersensitivity, and dysregulated neural signaling and/or central processing.2 Despite advancements in the understanding of IBS pathogenesis, diagnostic and therapeutic IBS biomarkers are limited.
Accumulating evidence suggests that gastrointestinal (GI) microbiome may be related to risk of IBS, but may also mediate many of the mechanisms that underlie symptoms including motility,3, 4 permeability,5 immune activation,6, 7 communication along the brain-gut axis,8 and visceral sensation.9 Characterization of microbial composition in IBS has suggested decreased microbial diversity, reduced temporal stability, or changes in the relative abundance of specific bacteria in patients with IBS.10, 11 However, these findings have not been sufficiently consistent across studies to establish a clear microbial profile in IBS and gaps in our understanding of the functional microbiome persist. It is now evident that complementary strategies of microbial metabolites will be crucial to gathering insights into the impact of the microbiome in IBS.
Atypical profiles of microbial metabolites including luminal bile acids12, 13 and short chain fatty acids (SCFA) have been described in some patients with IBS.14, 15 Bile acid malabsorption (BAM) is recognized as a common mechanistic IBS subtype,13, 16 and may be associated with physiological traits, symptoms, and quality of life.17–20 Recently, researchers have examined microbial contributions to BAM in IBS to report alterations such as enrichment of Clostridia bacteria including C. scindens,21 lower microbial alpha diversity, higher Firmicutes to Bacteroidetes ratio,20 and presence of endoscopically visible biofilms correlating with overgrowth of Escherichia coli and Ruminococcus gnavus.22 SCFA are produced by anaerobic fermentation of dietary fibers and resistant starch that enter the colon and regulate intestinal homeostasis and physiology.23 Compared to bile acids, the role of SCFA in IBS is less well understood. Studies of stool SCFA in IBS have yielded variable results, which may be related to the heterogeneity of the IBS patient populations and multiple pathways through which SCFA may modulate intestinal physiology. Therefore, while stool SCFA are unlikely to serve as categorical IBS biomarkers, they may provide critical insights into the pathophysiological mechanisms that underlie IBS symptoms. Accordingly, studies10, 15, 24 that have assessed stool SCFA in distinct IBS subgroups have reported more consistent associations of stool SCFA with IBS subtypes as well as correlations of stool SCFA with measurable IBS traits such as colonic transit, bowel functions, and bile acid excretion.24–27 Despite these reports, the intercorrelation between SCFA, bile acids, and transit time complicates the assessment of whether microbial composition is directly responsible for SCFA profiles or if relationships between the intestinal microbiome and excreted SCFA is clinically valuable due to the complex and dynamic nature of the microbial ecosystem. However, recent work28 has suggested that individual taxa, rather than complex ecological communities, could drive changes in SCFA output in response to dietary fiber. Therefore, to address these questions, we conducted an in-depth investigation of GI microbiome structure and function, stool SCFA, and IBS endophenotypes (transit, bile acids, bowel functions) in adults with and without IBS.
MATERIALS AND METHODS
Participant Recruitment and Study Design
The study was approved by the Indiana University Institutional Review Board and the protocol registered within ClinicalTrials.gov (NCT02981888). The study was designed as an observational investigation of stool SCFA, stool bile acids, colonic transit, and stool microbiota in adults with and without IBS. We enrolled adults ages 18-65 years of age through the Indiana University Gastroenterology Clinics, the Indiana Clinical and Translational Research Institute Research Registry, and from the local community. We included individuals with IBS with diarrhea (IBS-D) or IBS with constipation (IBS-C) according to Rome IV criteria29 and healthy controls with no prior history of GI diseases or symptoms. Detailed eligibility criteria are available in the Supplemental Methods.
Data Collection
Study eligibility, medications, medical history, and baseline diet using a food frequency questionnaire30were assessed during a screening visit with a study physician. Over a two-week period, data were collected on bowel functions using a standardized bowel pattern diary including the Bristol stool form scale.31 All participants submitted a 48-hour stool collection collected during the last 2 days of a 4-day 100 g fat diet, consistent with previously validated methods for measuring of stool bile acids using home kits. Specimens were refrigerated during the collection period, returned to the research team on ice, and stored at -80οC.
Colonic Transit by Radiopaque Markers
Participants underwent assessment of colonic transit time with a previously validated and optimized method using radiopaque markers.32, 33
Stool SCFA and Bile Acids
Frozen aliquots of stool were shipped to the Metabolite Profiling Facility at Purdue University to measure total and individual (acetate, propionate, butyrate) SCFA concentrations per mg of dry weight by liquid chromatography-mass spectrometry (LC-MS) using published methods34 and to the Mayo Clinic Department of Laboratory Medicine and Pathology to measure total and primary stool bile acid levels by high-performance LC-MS through a commercially available, CLIA-approved assay.17, 35, 36
DNA Extraction, Purification, and Sequencing
Genomic DNA was isolated from stool using the QIAmp® PowerFecal® DNA kit (QIAGEN Inc., Germantown, MD, USA). DNA quality and concentration were on a Qubit fluorometer. Purified DNA underwent library preparation (Nextera XT, Illumina) and paired-end (2×150 bp) sequencing using the NovaSeq v1.5 SP (Illumina, San Diego, CA, USA) to target a sequencing depth of 40M sequences per sample.
Metagenomic Data Analysis
Metagenomic sequencing reads were quality filtered and processed for taxonomic profiling using both MetaPhlAn3. Additive log-ratio (ALR) transformation was applied to analyze differential taxonomic abundances. Based on the identified microbial taxa from each sample, β-diversity indices (e.g., Bray-Curtis dissimilarity) were calculated using the R packages phyloseq and vegan.37, 38
Statistical Considerations
We summarized major endpoints of interest: (1) stool microbiome composition, (2) stool SCFA concentrations (total, acetate, propionate, butyrate), total and percent primary stool bile acids, and (3) colonic transit time (overall and segmental). Our primary objective was to examine the correlation between the stool microbiome and SCFA l (microbe-SCFA associations) across clinical groups (IBS-D, IBS-C, healthy controls) after accounting for transit and bile acids and to determine if microbe-SCFA relationships vary across the IBS spectrum.
We compared microbial taxa abundances between groups and analyzed associations of taxa abundances with quantitatively defined endophenotypes (stool SCFA, stool bile acids, and transit) for the collective cohort and within groups. Associations of microbiome composition with endpoints of interest were assessed using general linear regression models (GLM) adjusted for covariates including age, sex, and BMI. Only high abundant species (relative abundance ≥0.1%) that were prevalent in ≥2 specimens within ≥1 clinical group were considered. In patients with IBS-D, we analyzed multivariable associations of microbiome composition between patients with and without BAM.
Functional profiling was conducted using the HUMAnN3 pipeline annotated by KEGG (Kyoto Encyclopedia of Genes and Genomes) orthogroups (KO). Relative abundances were used to examine the associations of gene family/pathway abundances across clinical cohorts. A manually curated gut-metabolic analysis framework39 was applied to examine KO identifiers associated with carbohydrate degradation, SCFA production or metabolism, and mucin degradation using GLM adjusting for covariates. As stool form40 has been identified as a strong source of human microbiota variation and closely linked to transit,41, 42 we further explored associations of functional potential across groups in a subset of participants who were selected based on stool form features (i.e. those individuals best representing the stool types within their respective clinical group according to bowel diary data) using the Kruskal-Wallis test. For example, we chose the IBS-D and IBS-C participants with the loosest and firmest stool types, respectively, and controls with consistently normal stool types. For all endpoints, missing values were excluded from the analysis for that endpoint.
RESULTS
Participant Characteristics
Among 96 volunteers who underwent screening evaluation, 71 completed the study, and 58 participants (Supplemental Figure 1) with a mean [±SD] age = 35.5 (±13.8) years and mean [±SD] BMI = 26.2 [±7.5] kg/m2) were included in the final analysis after excluding those who ineligible (n=16), lost to follow-up (n=9), or and/or did not provide SCFA data (n=13). Baseline clinical characteristics (Table 1) were not significantly different across groups (all p=ns) Comparisons of quantitative traits demonstrated differences in total stool bile acids and transit between IBS-D and control participants, and in stool SCFA (total, acetate) between IBS and controls. (Table 2).
Clinical Characteristics of Patients with Irritable Bowel Syndrome (IBS) and Controls
Quantitative Traits in Patients with Irritable Bowel Syndrome (IBS) and Controls
Stool microbiome composition differs between IBS and health and between IBS subtypes
Shotgun metagenomic sequencing of stool samples was undertaken to obtain total of 3.1 Gb of sequence data after removal of contaminants with an average of 37.6 million paired-end reads per sample (deposited into NCBI with accession number SUB13882354). Taxonomic classification identified 461 taxa at species level classification. Comparisons of microbial metagenomes based on Bray-Curtis Dissimilarity revealed significant divergence (Supplemental Figure 2) of community distance between groups (p=0.003, R2 =0.06) after adjusting for age, BMI, and diet. We compared abundances of individual taxa across groups using the Kruskal-Wallis test to identify 18 unique and differentially abundant taxa. Highest mean relative abundances of 14 taxa (Supplemental Table 1) including Dorea sp. CAG:317, Blautia sp. CAG:257, Ruminococcus gnavus, and Proteobacteria bacterium CAG:139 were observed in IBS-D, which have previously been linked to mechanisms such as BAM21, 22 and serotonin biosynthesis in patients with IBS-D.43 Lawsonibacter asaccharolyticus, a butyrate-producing bacterium,44 abundance was highest in IBS-C and Firmicutes bacterium CAG:83 abundance was highest in controls.
We followed unadjusted analyses with pairwise comparisons of ALR-transformed taxa abundances using covariate-adjusted GLM, focusing on high abundant species, to demonstrate significant differences in in 17 pairwise comparisons including 12 unique species (Figure 2) of which 11 exhibited differential abundances of ≥3-fold. Among these 11 taxa, we observed significantly higher abundances of Dorea sp. CAG:317 and Bifidobacterium pseudocatenulatum in IBS-D compared to IBS-C or controls and higher abundances of Blautia sp. CAG:257, and Proteobacteria bacterium CAG:139 in IBS-D compared to controls. We found significantly higher abundances of Clostridium sp. CAG:58 and lower abundances of Firmicutes bacterium CAG:83 in both IBS-D and IBS-C relative to controls, respectively. Abundances of Akkermansia muciniphila and Prevotella copri, were increased in IBS-C compared to controls. A modest (≤3-fold) increase in abundance of Monoglobus pectinilyticus, a pectin-degrading specialist,45 was observed in IBS-C compared to controls or IBS-D. Differences of ≥3-fold in ALR-transformed abundances among high abundant bacteria were also observed in 151 pairwise comparisons (Supplemental Table 2), but were not statistically significant after adjustment for covariates.

Mean percent abundances (standard deviation) of significantly different bacterial taxa with ≥3-fold differences in pairwise comparisons of clinical cohorts.

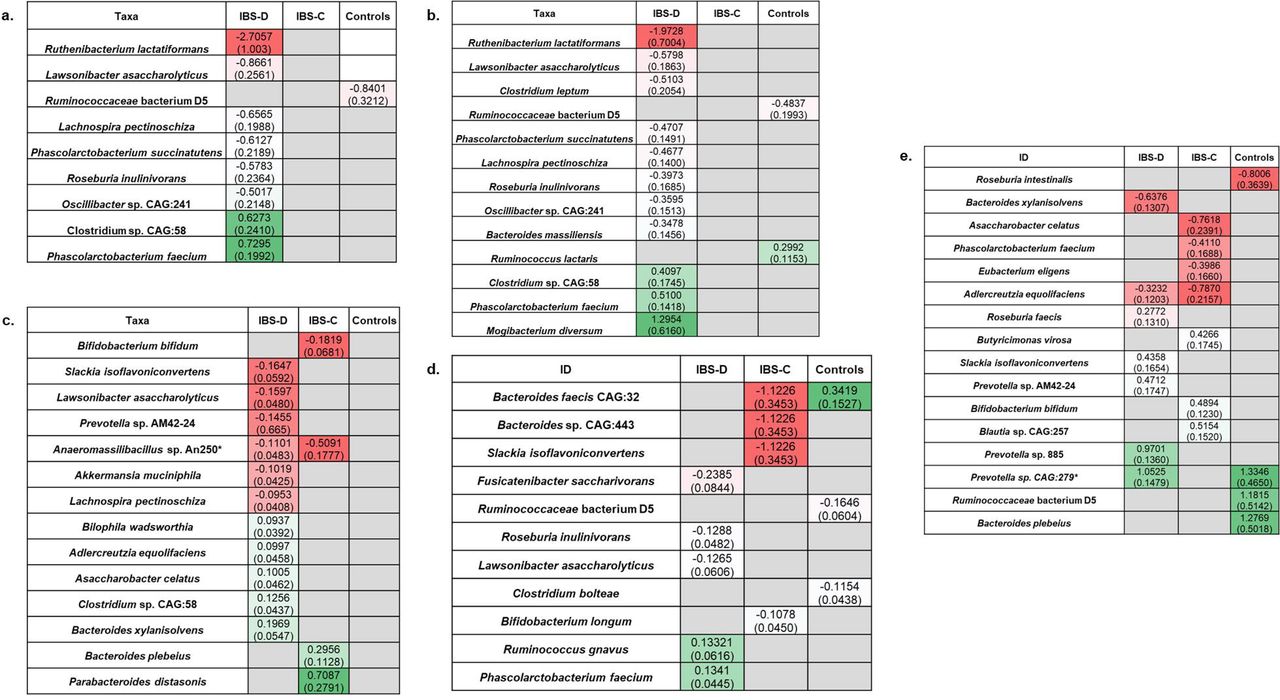
Significant associations of bacterial taxa with short chain fatty acids (SCFA). Panels show associations with total SCFA [a], acetate [b], butyrate [c], propionate [d], and acetate to butyrate ratio [e] within cohorts. Figure is presented as a heatmap demonstrating regression coefficient estimates (standard error) with red indicating negative correlations and green indicating positive correlations.
Microbe-SCFA associations differ between IBS subtypes and controls
We analyzed associations of taxa abundances with stool SCFA, bile acids, and transit using multivariable GLM, focusing on microbe-SCFA associations that did not overlap with other microbe-endophenotype (i.e., transit or bile acids) associations and assessed if relationships were maintained or differed across clinical groups.
Unique microbe-SCFA associations were observed for five high abundant taxa in the collective cohort; these taxa were not associated with transit (overall or segmental) or bile acids (total or % primary) and included species known to be involved in SCFA production and fiber fermentation such as Agathobaculum butyriciproducens.46 We identified eight unique microbe-SCFA associations that were specific to IBS-D. These associations involved several bacterial species including L. asaccharolyticus, Phascolarctobacterium faecium, Lachnospira pectinoschiza, and Roseburia inulinivorans with known capacity for SCFA production,44 fiber fermentation,47 and mucin degradation.48 One unique microbe-SCFA association was observed in controls (Ruminococcaceae bacterium D5). Another unique microbe-SFCA association was observed in patients with IBS-C (Christensenella minuta), but this bacterial species did not meet the high abundant threshold in any individual group. No unique microbe-SCFA associations were shared across groups (Figure 3a).

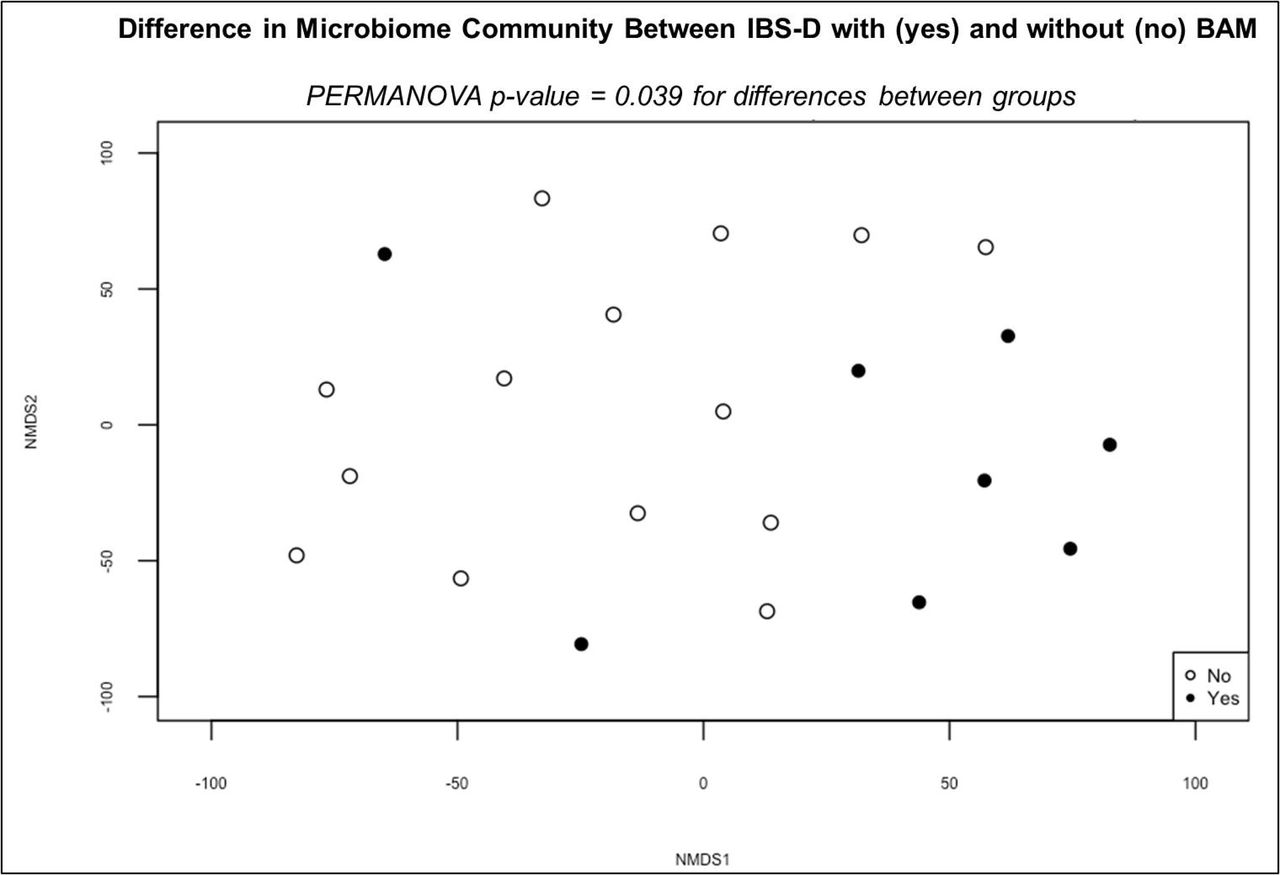
Non-metric multidimensional scaling (NMDS) representation of beta diversity in patients with (yes) and without (no) bile acid malabsorption (BAM) using taxonomic classification from MetaPhlAn.
Keeping in mind SCFA effects may depend on SCFA type, we investigated microbe-SCFA relationships by SCFA type (acetate, butyrate, propionate) and by acetate to butyrate ratio. We found unique associations of taxa abundances with acetate, butyrate, propionate and acetate to butyrate ratio that were both shared and distinct across groups, though most were distinct (Figure 3b-e). Most unique microbe-SCFA associations were observed in IBS-D. In IBS-D, abundances of 10 taxa including known SCFA producers (e.g., Ruthenibacterium lactatiformans,49 L.asaccharolyticus, P. faecium)50 fiber fermenters (e.g. Clostridium leptum47), β-fructan utilizers (e.g. R. inulinivorans47, 51) and host mucin-degrading species (Bacteroides massiliensis52, 53) were significantly associated with stool acetate. Eight of the 10 were negatively correlated with stool acetate in IBS-D. No microbe-acetate associations were shared across groups. Ten high abundant taxa were significantly associated with stool butyrate concentrations in IBS-D (Figure 3c), but not with transit or bile acids; five species including L. asaccharolyticus, A. muciniphila, and L. pectinoschiza demonstrated negative associations with butyrate concentrations. Anaeromassilibacillus sp. An250 abundance was negatively associated and butyrate in both IBS-C (p=0.02) and IBS-D (p=0.03); however, relative abundance levels were less than 0.1%. No other microbe-butyrate associations were shared. Within clinical groups, we identified unique associations between taxa abundances and propionate. Five high abundant bacteria (Figure 3d) were negatively (Fusicatenibacter saccharivorans, R. inulinivorans, L. asaccharolyticus) and positively (R. gnavus, and P. faecium) associated with stool propionate in IBS-D. Several high abundant taxa were specifically associated with acetate to butyrate ratio within clinical groups (Figure 3e). A negative association between Adlercreutzia equolifaciens abundance and acetate to butyrate ratio was shared between IBS-D and IBS-C. A positive association between Prevotella sp. CAG:279 abundance and acetate to butyrate ratio was shared between IBS-D and controls, although relative abundance levels did not meet the high abundance threshold in any clinical cohort. None were shared between patients with IBS-C and controls.
Relationships of SCFA-associated microbes and bile acid malabsorption (BAM)
Stool microbial metagenomes were analyzed in 22 IBS-D patients with (n=8) and without (n=14) BAM. Comparisons of beta diversity based on Euclidian dissimilarity of ALR transformed abundance data showed significant dissimilarity (p = 0.039; R2 = 0.061) between patients with and without BAM (Figure 4). Significant differences in six high abundant species were observed between groups, including L. asaccharolyticus R. inulinivorans, L. pectinoschiza, and F. saccharivorans which were also associated with stool SCFA, but not bile acids, in IBS-D. These four species with known SCFA-producing capacity were negatively correlated with BAM.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Significant differences in functional potential for carbohydrate degradation, short chain fatty acid production or metabolism, and mucin degradation across clinical cohorts in representative subset selected by stool form characteristics.
Functional potential for carbohydrate degradation, SCFA metabolism, and mucin degradation
Examination of microbial gene families and pathways yielded 949,223 total gene families within the metagenome data set including 4,042 named KO terms. We applied the gut metabolic module (GMM) framework to analyze modules related to carbohydrate degradation, SCFA production or metabolism including bacterial cross-feeding pathways, mucin degradation, and one module related to serine degradation based on significant species-based predictors. We identified significant associations of functional microbiome features with clinical-and SCFA-phenotypes. Abundances of KO identifiers within modules for lactose degradation, (p=0.0026), serine degradation (p=0.019), and propionate production (p=0.026) were significantly enriched in IBS-C compared to controls. Relative abundance of a KO identifier within a galactose degradation module was significantly reduced in IBS-D (p=0.033) compared to controls. In the collective cohort, we applied the GMM framework to analyze the relationships between metagenomically-encoded functions and SCFA-endophenotype. We identified 23, 23, 15, and 16 KO identifiers from relevant GMMs that were significantly associated with total SCFA, acetate, butyrate, and propionate, respectively. Within a representative subset (n=6 controls, n=5 IBS-D, n=5 IBS-C) selected by stool form characteristics, functional metagenomic analyses demonstrated that microbial genes/pathways associated with SCFA production/metabolism and degradation of carbohydrates and mucin were differentially associated with clinical cohort (Figure 5).
DISCUSSION
It has been proposed that changes in intestinal SCFAs in some patients with IBS are caused by shifts in microbial structure and function to drive and maintain symptoms. We hypothesized that specific features of the GI microbiome may correlate with SCFA output and that these relationships may distinguish IBS subtypes and endophenotypes. We further hypothesized that functional features of the GI microbiome defined on by substrate preferences and SCFA metabolism may be linked to bowel dysfunction. Using a dual-omics approach, our findings demonstrate microbiota-SCFA relationships differ between IBS subtypes and endophenotypes, and between patients with IBS and healthy controls. Our findings suggest that key taxa may predict IBS mechanisms and that the functional repertoire of the intestinal microbiome could influence bowel functions. By capturing quantifiable traits, we also gather evidence suggesting the microbiome has direct effects on excreted SCFA and identify microbe-SCFA relationships that are not solely explained abnormal transit or bile acid excretion.
While numerous studies have reported compositional changes10, 11 in IBS such as decreased microbial diversity and altered abundances of individual taxa, consistent patterns are difficult to pinpoint. Recent data suggest that putative microbial biomarkers are related to mechanistic and host-specific features in IBS rather than the overall clinical syndrome. Consistent with these prior studies, we observed differences in overall microbial community composition across groups. Comparisons of taxa abundances demonstrated significant changes were most common in IBS-D and predominantly characterized by higher abundances of several bacterial species including Blautia sp. CAG:257, R. gnavus, Dorea sp. CAG:317, and Proteobacteria bacterium CAG:139. R. gnavus, a Lachnospiraceae, has previously been identified to play a pathogenic role in IBS-D through serotonin biosynthesis,43 biofilm formation and increased stool bile acid excretion,22 production of proinflammatory polysaccharides,54 and mucin degradation.55 Interestingly, the relationship between R. gnavus and IBS lost its significant in adjusted analyses due to a positive association between R. gnavus and BMI which may suggest a role R. gnavus in explaining the connection between IBS-D or chronic diarrhea with metabolic syndrome and obesity-related disorders.56, 57 Associations of IBS-D with other taxa including Dorea sp. CAG:317, Blautia sp. CAG:257, and Proteobacteria bacterium CAG:139 remained and a new association between B. pseudocatenulatum and IBS-D emerged in multivariable analysis after covariate adjustment. Mucin degradation has been described among several Dorea species and the genus Dorea belongs to the Lachnospiraceae family, a major producer of SCFA58 while B. pseudocatenulatum is metabolically adapted for utilization of dietary carbohydrates including long-chain xylans.59 Recently Jacobs et al.60 reported significant increases in transcript abundances for fructooligosaccharide and polyol utilization and upregulation of transcripts for fructose and glucan metabolism as well as carbohydrate fermentation in IBS-D relative IBS-C, suggesting differences in microbial metabolism between IBS subtypes. In our study, only two bacterial species, A. muciniphila and P. copri were significantly increased in IBS-C. Previous studies have reported increased A. muciniphila in IBS-C to be protective against experimentally-induced colitis in mice and a positive association between mucosal P. copri abundance and abdominal pain in patients with IBS without prior history of infection.61, 62 Considered alongside previously published work, our findings may suggest that while distinct changes in microbiome composition occur in IBS-D vs. IBS-C, microbial dysbiosis or changes in microbial metabolism could play a larger role in IBS-D than IBS-C. This observation may also explain the higher prevalence of mixed- and diarrhea-predominant symptoms in post-infection IBS as well as the limited evidence63 supporting the efficacy of antimicrobial treatments such as rifaximin in IBS-C.
We have previously shown that transit and BAM correlate with SCFA excretion in IBS.27 Others have reported that microbiome associations with transit are largely influenced by altered bile acid biotransformation.19 In this current study, several bacterial species with fermentative capacity or varied substrate processing functions correlated with total stool SCFA, but not with transit or bile acids. Most associations were observed in IBS-D, suggesting that microbial substrate utilization patterns are particularly important in IBS-D. Interestingly, negative correlations were observed between several SCFA-producing species including R. inulinivorans and total SCFA. While R. inulinivorans is a known fiber fermenter,47 a previous study R. inulinivorans possesses the ability to forage mucin through a metabolic interplay with other members of the intestinal microbiota 48 suggested that and is known for net acetate uptake,64 which may explain its significance in IBS-D and correlation with reduced SCFA. Analysis of the main SCFA types also demonstrated negative associations between acetate and abundances of butyrate-producing and mucin-degrading bacterial species in IBS-D. Future work should investigate the effects of bacterial cross-feeding, microbial conversions, and interactions of the colonic microbiome with diet- and host-derived substrates that may destabilize the microbiota-mucosal interface to expose vulnerabilities and promote pathophysiological mechanisms that drive IBS.
In our comparisons across IBS-D patients by with and without BAM, we observed BAM to be associated with reduced abundances of several SCFA-producing bacterial species. Similar observations have been made in studies linking biofilm formation to bile acid accumulation and reduced SCFA-producing bacteria.22, 65 Results suggest that BAM exerts indirect effects on the intestinal metabolome or the overall metabolic potential of the colonic microbiota characterized by a decline in SCFA-producing capacity.
A few bacterial species were associated with SCFA in more than one clinical cohort Adlercreutzia equolifaciens, which has been described as an anti-inflammatory commensal in non-alcoholic fatty liver disease,66 was negatively correlated with acetate to butyrate ratios in both IBS-D and IBS-C. Two other species also demonstrated relationships with SCFA across more than one group, although relative abundance levels were low overall. These species included Anaeromassilibacillus sp. An250, which was negatively correlated with stool butyrate in both IBS-D and IBS-C and Prevotella sp. CAG:279 which was positively correlated with acetate to butyrate ratios in both IBS-D and controls. Recently, Butler et al. reported enrichment of Anaeromassilibacillus sp. An25067 in patients with social anxiety disorder and preclinical studies have described reversal of anxiety-like behaviors with gut commensal-derived butyrate in mice.68 Taken together, these data could point towards Anaeromassilibacillus sp. An250 as a novel microbial biomarker of pathogenic mechanisms involving the microbiota-gut-brain axis, possibly through the effects of microbially-derived SCFA.
We also assessed of functional variations in the GI microbiome to reveal differential abundances of KO identifiers belonging to metabolic processes related to degradation of carbohydrates and serine as well as SCFA production. In a subset of participants selected by stool form characteristics, we identified changes in substrate degradation potential and fermentative capacity between IBS and controls and between IBS subtpes that were characterized by increased capacity for starch degradation in controls, enhancement of distinct mucin-degrading functions in IBS-D vs. IBS-C, and reduced pectin degrading potential in IBS-C. Our results may imply that abnormal stool form in IBS is influenced by microbiota-encoded substrate preferences.
Despite study strengths, we recognize the limitations of this work including cross-sectional sampling, which overlooks the fluctuating nature of the microbiome. To address this, we assessed relationships between microbial abundances and quantifiable IBS endophenotypes that were defined at the time of specimen collection while accounting for baseline covariates. All participants were instructed to consume a 4-day high fat diet while otherwise maintaining their usual diet. Previously, we examined the effect of both habitual diet and real-time intake on excreted stool SCFA to find that that modest variations in macronutrient intake including polysaccharides did not exert substantial effects on excreted SCFA. We enrolled patients with opposing IBS phenotypes from both the university clinics and the surrounding communities to increase our ability to detect differences between clinical cohorts and limit referral bias. We also acknowledge that causal microbial mechanisms cannot be determined through the current work. However, we applied a dual-omics approach to investigate biologically relevant changes in intestinal microbiome structure and function. Finally, we cannot discount the possibility we may have missed significant taxa due to the moderate sample size and a risk for Type I errors as we did not adjust for false discovery rate in our analysis of taxonomic differences. However, our analytic approach was based on endpoints that were determined a priori and we applied a conservative strategy by focusing on taxa that were associated with SCFA, but not with bile acids or transit. We further limited analyses to high abundant taxa and to species that exhibited >3-fold change in abundance while focusing on the biological plausibility of our results. Findings from this study should be validated in larger, longitudinal cohorts, but provide a framework that may be used to define key microbial interactions and targets in IBS.
In conclusion, main findings from this study highlight microbiota-SCFA patterns vary across clinical IBS phenotypes and endophenotypes. Prominent shifts in microbial structure are observed in IBS-D and appear to affect metabolic capacity, substrate preferences, and metabolite profiles. Altered microbial substrate uptake may impact stool form or bowel function in IBS. Collective, these findings may suggest putative microbial biomarkers or metabolically influential features that could be targeted for future development of microbiota-based therapeutics in IBS.
Data Availability
Microbiome sequencing reads are deposited into NCBI with accession number SUB13882354. The data that support the findings of this study are available upon reasonable request from the corresponding author.
Funding
AS is supported by NIDDK K23DK122015 and R03DK132446.
Disclosures
AS serves on Ardelyx Scientific Communications Advisory Board for irritable bowel syndrome with constipation.
Author Contributions
Developing study concept: AS. Planning study design: AS, HX, XG. Participant recruitment: AS, MRW, RS, TJS, NR, MB JW, CL, AG, JK, RA. Data collection and study procedures: AS, MRW, CL, AG, JK, ET. Data Management: AS, YX, MRW, CL, JK, HX, XG. Data Analysis and Interpretation: YX, XG. Drafting the manuscript: AS, XG. Critically revising the manuscript: MRW, RS, TJS, NR, MB, JW, ET, HX.
Data Transparency Statement
Microbiome sequencing reads are deposited into NCBI with accession number SUB13882354. The data that support the findings of this study are available upon reasonable request from the corresponding author.
Acknowledgements
The authors would like to thank the Mayo Clinic Department of Laboratory Medicine and Pathology and Purdue University Metabolite Profiling Facility for their collaboration and assistance with quantification of stool bile acids and short chain fatty acids.
Abbreviations
- ALR
- additive log-ratio
- BAM
- bile acid malabsorption
- CTT
- colonic transit time
- gDNA
- genomic DNA
- GI
- gastrointestinal
- GLM
- generalized linear model
- GMM
- gut metabolic module
- IBS
- irritable bowel syndrome
- IBS-D
- IBS with diarrhea
- IBS-C
- IBS with constipation
- KEGG
- Kyoto Encyclopedia of Genes and Genomics
- KO
- KEGG Orthogroup
- LC-MS
- liquid chromatography-mass spectrometry
- SCFA
- short chain fatty acids
References



