
Abstract
Alzheimer’s disease affects our cognitive neurophysiology by loss of neurones, synapses and neurotransmitters. An improved mechanistic understanding of the human disease will facilitate new treatments. To this end, biophysically-informed dynamic causal models can support inferences around laminar and cell-specific disease effects from human non-invasive imaging. Based on pre-clinical models and effects of cholinesterase inhibitors, we hypothesised that Alzheimer’s disease would affect the modulation of superficial pyramidal cell gain and extrinsic connectivity between pyramidal cells of different regions in hierarchical cognitive networks. Magnetoencephalography (MEG) was recorded during an auditory mismatch negativity task from healthy adults (n=14) and people with symptomatic Alzheimer’s disease or mild cognitive impairment (n=45, all amyloid-biomarker positive) at baseline and after 16 months. Fourteen people from the symptomatic group had repeat magnetoencephalography at two weeks to assess test-retest reliability. Sensor-level data were analysed using t-tests of the mismatch negativity amplitude from 140ms to 160ms. The repetition effect was assessed with repeated-measures analysis of covariance, using the average evoked response in the mismatch negativity time window as the repeated measure. An absolute, intraclass correlation model of the test-retest data assessed mismatch negativity amplitude reliability. We then fitted dynamic causal models to the evoked responses over 500ms. Second-level parametric empirical Bayes across participants examined the effect of (1) group, patients vs controls, and (2) progression, baseline vs follow-up, on the model parameters reflecting pyramidal cell gain modulation and extrinsic connectivity. There was a significant effect of both disease and progression on the mismatch negativity amplitude (patients vs controls, T=-1.80, p=0.04; patient baseline vs follow-up, T=-2.72, p=.005), which had excellent reliability (ICC=0.95, p<.001). Parametric empirical Bayes revealed strong evidence (posterior probability>95%) that Alzheimer’s disease reduced extrinsic connectivity and superficial pyramidal cell gain modulation, which was reduced further at follow up assessment. The mechanistic modelling confirmed the hypothesis that reduced superficial pyramidal cell gain modulation and extrinsic connectivity can explain the observed neurophysiological effect of Alzheimer’s disease. This approach to non-invasive magnetoencephalography data may be used for experimental medicine studies of candidate treatments, and bridge clinical to preclinical models of drug efficacy.
Introduction
Developing effective treatments for Alzheimer’s disease remains a major challenge (Anderson et al., 2017). Effective treatments should target the disease mechanisms behind the neurophysiological and cognitive deficits (Yiannopoulou et al., 2019). To develop these treatments in vivo, human assays that quantify the causes of neurophysiological change are needed (Drummond and Wisniewski, 2017). Here, we test the sensitivity of the inversion between magnetoencephalography and generative models of human neurophysiology to the presence and progression of Alzheimer’s disease. This approach allows human, in vivo characterisation of neural dynamics by cortical layer and cell type from non-invasive neuroimaging (Friston et al., 2003).
We focus on the impact of Alzheimer’s disease on the connectivity of pyramidal cells. Pyramidal cells in superficial layers of cortex are modulated by currently licenced cholinergic treatments for dementia (Moran et al., 2013). For example, galantamine modulates activity of superficial pyramidal cells, leading to changes in neurophysiological responses to rapidly changing stimuli. In rodent models, cholinergic agonists and antagonists have opposing dose-dependent effects on superficial pyramidal cell gain modulation (Schöbi et al., 2021). The hippocampus, which has early and significant involvement in the course of Alzheimer’s disease, may be involved in inhibiting and disinhibiting superficial pyramidal cells through regulation of acetylcholine (Barron et al., 2020).
Pyramidal cells are directly affected by the molecular hallmarks of Alzheimer’s disease: extracellular beta-amyloid plaques (Braak and Braak, 1997) and intraneuronal tau tangles (Braak and Braak, 1991; Thangavel et al., 2008). Tau aggregates develop in pyramidal cells and can spread between pyramidal cells (Braak and Tredici, 2018). Variance in the distribution and burden of tauopathy in Alzheimer’s disease is closely associated with the clinical phenotype (Ossenkoppele et al., 2016), cognitive deficit (Nelson et al., 2012) and rate of cognitive decline (Malpetti et al., 2020; Tanner et al., 2022). The effect of aggregated tau on cognition in part results from synaptic toxicity and dendritic dearborization of pyramidal cells (Allard et al., 2012; Merino-Serrais et al., 2013; Mijalkov et al., 2021).
Identification of pyramidal cell function in humans, in vivo, can be achieved by invasive electrocorticography, usually in people undergoing evaluation of epilepsy. This is not practical as a basis for research and drug development in the context of Alzheimer’s disease. An alternative approach is the inversion between functional neuroimaging (e.g., electro- and magneto-encephalography, E/MEG) and biophysical models of the cortex. This approach has been used to study the effects of anti-NMDA autoimmune encephalopathy (Symmonds et al., 2018), inherited channelopathies (Gilbert et al., 2016), ageing (Moran et al., 2014), pathophysiology of non-Alzheimer dementias (Shaw et al., 2019; Adams et al., 2020, 2021, 2023) and confirmatory studies of drug interventions (Moran et al., 2013; Adams et al., 2021).
Here we use generative models with non-invasive human MEG to test the hypothesis that Alzheimer’s disease affects pyramidal cell gain and connectivity, which influence the neurophysiological response to unexpected inputs. We do so in the context of a sensory mismatch negativity paradigm, in which participants automatically learn and adapt to changing auditory stimulus frequency. This learning reflects short-term plasticity (Garrido et al., 2009) and is achieved by iterative updating of top-down predictions by bottom-up prediction errors (Auksztulewicz and Friston, 2016). As top-down predictions of a repeated stimulus become more accurate with short-term learning, prediction errors (and their neurophysiological correlate) reduce. With repetition, the network becomes more precise in its predictions such that prediction errors acquire greater precision (Auksztulewicz and Friston, 2016). An initial deviant stimulus retains the high precision of the previous standard, thereby enhancing the signal of the large prediction error (i.e., a large magnitude error made with high precision). Following such a large prediction error, the precision (c.f., the confidence, in lay terms) in the model is reduced, such that the subsequent prediction error is less precise and generates an attenuated mismatch response.
The precision of prediction errors is proposed to be encoded by the gain modulation of superficial pyramidal cells (Moran et al., 2013; Schöbi et al., 2021), while prediction error is proposed to be mediated by superficial pyramidal cell forward connections (Auksztulewicz and Friston, 2016). In Alzheimer’s disease, this mechanism is disrupted with changes including reductions in precision and prediction error signalling (Kocagoncu et al., 2021). Indeed, novelty encoding (Bastin et al., 2019) and auditory predictions (Pérez-González et al., 2022) are disrupted in Alzheimer’s disease, particularly in parietal and medial temporal cortices (Billette et al., 2022; Pérez-González et al., 2022). This deficit would not only impair learning, but also signal processing and perception in a noisy environment, as observed in Alzheimer’s disease (Pérez-González et al., 2022).
Our overarching hypothesis is that the observed physiological effect of Alzheimer’s disease on the mismatch negativity response is explained by reduced gain modulation of superficial pyramidal cells and reduced effectiveness of extrinsic connectivity between pyramidal cell populations of connected regions. We test two specific predictions: (1) symptomatic Alzheimer’s disease, in the form of amyloid-positive mild cognitive impairment and mild Alzheimer-type dementia, is associated with reduced gain modulation of superficial pyramidal cells and reduced extrinsic connectivity between pyramidal cells; (2) the same features of the cortical microcircuit change with disease progression.
Materials and methods
Participants
Data were drawn from the New Therapeutics in Alzheimer’s Disease (NTAD) multicentre longitudinal observational study (Lanskey et al., 2022). The study included neurologically-normal, control participants and participants with a clinical diagnosis of Alzheimer’s disease or mild cognitive impairment. Participants completed (i) amyloid screening through either positron emission tomography or cerebrospinal fluid collection, a positive amyloid status enabled study participation for prospective patient participants; (ii) a battery of clinical and neuropsychological assessments; and (iii) neuroimaging, comprising MRI and E/MEG protocols. People in the control group completed screening and baseline assessments only, while people in the patient group completed additional, longitudinal assessments as follows: clinical and neuropsychological assessments were completed at three timepoints with 12-16 months between sessions; blood collection, E/MEG and MRI were completed at baseline and again at 12-16 months. An additional E/MEG scan was completed two-weeks after baseline for a subset of people from the patient group (see Lanskey et al., 2022 for further details).
For the current analysis, we used the MEG, MRI and demographic baseline data from participants who completed the mismatch negativity MEG recording. Of the 50 people with mild cognitive impairment or Alzheimer’s disease who completed the NTAD baseline MEG scan at the Cambridge site, we excluded two people who did not complete the mismatch negativity task as the earphones did not fit comfortably, two people whose diagnosis was revised during follow-up and one due to data recording technical issues at baseline. Of the 15 people with mild cognitive impairment or Alzheimer’s disease who completed the test-retest MEG scans at the Cambridge site, one person was excluded from the analysis because of trigger-failures on the retest session. One control participant was amyloid-positive and excluded from the current analysis. The resulting participant numbers comprised 45 people with Alzheimer’s disease or mild cognitive impairment with a positive amyloid status and 14 neurologically-normal control participants. For the test-retest analysis, we used the subset of 14 participants from the test-retest subgroup of participants with Alzheimer’s disease or mild cognitive impairment. Clinical and cognitive data for participants are given in Table 1. Independent Student’s t-tests and Bayesian t-tests were used for continuous data, or chi-squared tests for categorical data, to identify differences at baseline between participants with Alzheimer’s disease and control participants.
Values are given as mean (standard deviation)
*denotes significant difference between baseline groups by frequentist and/or Bayesian statistics, as per text. Yrs, years; MMSE, mini-mental state examination; ACE-R, Addenbrooke’s cognitive examination revised; HIS, Hachinski ischaemic score; PET, positron emission tomography; SUVR, standardised uptake value ratio; CSF, cerebrospinal fluid
Task
We used a passive roving mismatch negativity task (Adams et al., 2020) while the participants passively watched a muted movie. The auditory mismatch negativity task elicits responses to unexpected ‘oddball’ stimuli followed by rapid plasticity as predictions are updated upon repetition of the new stimulus (Friston, 2005; Kiebel et al., 2007). Through non-magnetic earpieces, the participants hear blocks of short sinusoidal tones binaurally in phase, 60dB above the average auditory threshold. The tones are presented for 100ms at 500ms intervals with frequencies in the range 400 to 800Hz varying in 50Hz steps. The frequency of successive tones is the same within blocks but changes between blocks. The number of repeated tones per block varied from 3 to 11. Consequently, the first tone of each block represents a deviant tone, which becomes a new standard tone upon repetition.
Imaging acquisition
MEG data were collected on the ElektaVectorView system and MEGIN Triux Neo scanner. Both scanners were configured with 204 planar gradiometers and 102 magnetometers. The position of five head position indicator coils, standard fiducial points and over 300 additional head points were recorded using the Polhemus digitization system. ECG data was recorded by an electrode on the right clavicle and a second electrode on the left, lower rib. A reference electrode recorded from the left side of the nose and the ground electrode was placed on the left cheek. Electrodes on bilateral canthi and below and above the left eye recorded electro-oculogram data.
T1-weighted MRI was recorded from each participant with a 3T Siemens PRISMA scanner using a magnetisation prepared rapid gradient echo (MPRAGE) sequence (echo time, 2.91ms; inversion time, 900ms; repetition time, 2300ms; flip angle, 9 degrees; voxel size, 1mm isotropic; slice thickness, 1mm; slice number, 176; acquisition matrix, 256×240).
Data analyses
Pre-processing
MaxFilter v2.2 software (Elekta Neuromag) was used on the raw data to automatically detect bad channels, perform temporal signal space separation, and correct for head movement. Independent component analysis was performed using the EEGLAB toolbox (Delorme and Makeig, 2004) to detect and remove artefactual components that correlated with EOG and ECG timeseries using cardiac spatial and normative blink templates. The residual timeseries were subsequently reconstructed for further preprocessing using SPM12 (r7771). Data were bandpass filtered between 0.01Hz and 40Hz and epoched from −100ms to 500ms relative to stimulus onset (0ms). OSL’s artefact rejection algorithm (github.com/OHBA-analysis/osl-core) was used to remove residual bad channels and trials. Robust averaging was used to average epochs for deviant and repeated trials, with conditions weighted separately. A final low-pass filter was applied to the data to correct for potential high frequencies introduced during robust averaging.
Sensor space analysis
For the sensor space analysis, planar gradiometers were combined by calculating their root mean square. We calculated the mean response across planar gradiometers for each subject, for each trial type, for each session separately (baseline, two-weeks and annual follow up). The repetition effect was assessed with a repeated-measures ANCOVA, using the average evoked response in the mismatch negativity time window (140-160ms, Moran et al., 2013) with tone repetition number (deviant, repetition 1, repetition 2, repetition 3, repetition 4 and repetition 5) as the repeated measure, group (controls versus patients) for the between-subject measure and age as the covariate. We also assessed the difference waveform for each subject; the response to the deviant tone was subtracted from the response to the first stimulus repetition. We calculated the average amplitude of this difference waveform from 140 to 160ms. An independent sample t-test was used to compare means of the mismatch negativity amplitude between groups. Group average waveforms were plotted after averaging difference waveforms across participants of each group.
For the longitudinal patient data, the difference in means between baseline and follow-up sessions was assessed using a t-test. The repetition effect was assessed with a repeated-measures ANOVA, using the average evoked response in the mismatch negativity time window (140-160ms, Moran et al., 2013) with tone repetition number (deviant, repetition 1, repetition 2, repetition 3, repetition 4 and repetition 5) and session (baseline versus follow-up) as repeated measures.
We estimated test-retest reliability of the baseline and two-week data using an absolute, intra-class correlation model (Koo and Li, 2016). With similar means, the intra-class correlation coefficient is numerically equivalent to the product moment correlation (estimated as Pearson’s correlation coefficient).
First level network modelling
We used dynamic causal modelling with laminar-specific parameters to determine the effect of Alzheimer’s disease on the neurophysiological generators. We used a canonical microcircuit model for evoked responses (Bastos et al., 2012; Pinotsis et al., 2013) comprising of four cell populations at each of 8 cortical regions (see Figure 2): bilateral primary auditory cortex (left MNI coordinates: −42, −22, 7; right MNI coordinates: 46, −14, 8), bilaterial superior temporal gyrus (left: −61, −32, +8; right: +59, −25, +8), bilateral inferior frontal gyrus (left: −46, +20, +8; right: +46, +20, +8), and bilateral inferior parietal cortex (left: −58, −27, +30; right: +59, −41, +30). Six of these regions are based on a series of prior studies with healthy adults and those with non-Alzheimer dementia (Garrido et al., 2008; Phillips et al., 2015; Rosch et al., 2019; Adams et al., 2021). We extended the network to include the inferior parietal cortex (Lappe et al., 2013), given the impact of Alzheimer’s disease on parietal cortex (Jacobs et al., 2012; Talwar et al., 2021) and recent evidence of mismatch negativity responses from parietal cortex (Lappe et al., 2013; Cope et al., 2022). We defined multiple, alternative, symmetric model architectures, each representing plausible alternative hierarchical relationships between regions and their connectivity to include the inferior parietal cortex (Figure 2). Given that higher regions send backward connections to, and receive forward connections from, lower regions in a hierarchical network, we specified forward and backward connections according to the following hierarchy as set out previously (Garrido et al., 2008; Phillips et al., 2015; Rosch et al., 2019; Adams et al., 2021): inferiFor frontal gyrus > superior temporal gyrus > primary auditory cortex. These connections were unchanging across models. All models included auditory input to primary auditory cortex and expectancy inputs to inferior frontal cortex (Phillips et al., 2015). Models 1 and 4 had no connections between frontal and parietal nodes. Models 2 and 5 had the inferior parietal cortex higher than the inferior frontal cortex in the cortical hierarchy. Models 3 and 6 had the inferior frontal cortex higher than parietal cortex in the cortical hierarchy. Models 1-3 had no expectancy input to parietal cortex, while models 4-6 had expectancy input to parietal cortex Figure 2B. The laminar asymmetry of forward vs backward connectivity is based on the canonical microcircuit model described by Bastos and colleagues (2012).
We modelled the first 6 repetitions of tones in the mismatch negativity paradigm to identify the effect of repetition on parameters from the first tone of each block (deviant) to the 6th tone (a new ‘standard’ for each block). In anticipation of short-term plasticity within the dynamic causal models, we modelled the changes to neural responses over successive tone repetitions as varying by (1) exponential decay or (2) exponential decay combined with phasic change (as used by Garrido and colleagues, 2009). The second set of models with both exponential and phasic change embody the hypothesis of predictive coding for sequential neural responses to stimuli, allowing extrinsic connections to show stimulus-specific adaptation with exponential decrease with repetition; while intrinsic connections show a phasic response, reducing after the deviant tone and recovering with subsequent tones thereby reflecting the precision of prediction error (Garrido et al., 2009; Rosch et al., 2019).
The neuronal parameters for each participant’s dynamic causal model were inferred from their observed response fields of gradiometers using a lead field informed by subject-specific T1 magnetic resonance images. This inversion uses the Bayesian variational Laplace method that minimizes the free energy of the model as a cost function to estimate the free energy of the model’s and posterior estimates of its parameters (Friston et al., 2003, 2007). Of the 14 controls and 45 patient participants included at baseline, one control and five patients had an inversion failure during model fitting and were excluded from the parametric empirical Bayes (PEB) analyses. For the longitudinal session, three patients failed to invert during model fitting and were excluded from the longitudinal parametric empirical Bayes analyses.
Bayesian model reduction was applied to each individual’s dynamic causal model to identify the most likely explanatory model for group differences (the “winning” model) from the model space. The model space included 12 canonical microcircuit models (Figure 2A), with 2 sets of the 6 alternative extrinsic connectivity architectures among eight regions; one set describing the effect over sequential tones with an exponential decay and the second set, as a combination of exponential and phasic basis functions (Garrido et al., 2009). The free energies of reduced models were averaged (i) across all participants at baseline and (ii) separately, across all baseline and longitudinal data for patient participants. The same model had the highest model evidence for both baseline and longitudinal datasets. This model with the highest model evidence was taken forward to PEB analysis to study group differences.
The effect of Alzheimer’s disease
Second-level analysis with PEB of the winning model examined how disease group (healthy control vs Alzheimer’s disease) affected the change in generative model of successive repetitions after a change in tone frequency. We ran two analyses with PEB. For both, the PEB design matrix comprised a unitary regressor for the group mean (ones) and a regressor for group membership (zeros for control participants and ones for people with Alzheimer’s disease or mild cognitive impairment).
The first PEB analysis tested whether individual differences in the gain of superficial pyramidal cells explains the group difference in the scalp response to the mismatch negativity task. The gain modulation parameter represents a lump sum of features such as potassium conductances and hyperpolarisation after an action potential, which act as an auto-regulation or self-inhibition of the cell population (rather than being a biological inhibitory connection per se; Moran et al., 2013). A second PEB analysis tested the effect of individual differences in the extrinsic connectivity between pyramidal cells between all regions (both forward and backward connections). For both PEB models, Bayesian model comparison and averaging was performed over a model space of all parameter combinations with hemispheric symmetry and the Bayesian model average results were plotted.
The effect of Alzheimer’s disease progression
For the analysis of longitudinal change, we performed two second-level analyses with PEB applied to baseline and annual follow-up data from patients. Both PEB models included a group mean regressor and a regressor specifying whether the scan was at baseline, given a value of 0, or at the annual follow-up, given a value of 1. A third regressor specified the time in years between the baseline and follow-up scans for each patient (0 for baseline scans and with scores from 0.8 to 2.2 for follow-up scans mean-centred and standardised). Two such PEB models were inverted to examine the impact of disease progression on the (i) gain modulation of superficial pyramidal cells and (ii) extrinsic connectivity between superficial and deep pyramidal cells. For each PEB model, we performed Bayesian model comparison and averaging over a reduced model space of only those parameters that had differentiated patients versus controls at baseline, and then the Bayesian model average results were plotted.
Data availability
The code used is available at https://github.com/jlansk/dcm_cmc_ntad. Anonymised (unlinked) raw data will be made available via Dementias Platform UK, subject to managed access conditions that protect participant confidentiality and conditions of consent.
Results
Demographic and clinical data are shown in Table 1. There were no significant differences between the groups in terms of sex (χ²=0.98, p=.32), handedness (t=1.86, p=.40), or Haschinski ischaemic score (χ²=5.12, p=.16). Patients were on average older than controls (t=-3.80, p=<.001, BF=72). As expected, there were group differences for the mini-mental state examination (t=4.75, p<.001, BF=1163), ACE-R (t=5.02, p<.001, BF=2722), and clinical dementia rating (χ²=55, p<.001). The baseline data of those completing two-week retest magnetoencephalography, and annual follow-up, were similar to the whole baseline group.
Sensor-level effects of Alzheimer’s disease
The amplitude of the mismatch response from 140 to 160ms was significantly reduced in patients compared to controls at baseline (T = −1.80, p = 0.04, Figure 1B upper panel). The repeated-measures ANCOVA found a significant main effect of repetition (F(5, 280) = 38.0, p<0.001); that interacted with group (F(5, 280) = 2.48, p = 0.03), where the responses were smaller for patients particularly for early repetitions.

(A) Amplitude of the evoked scalp responses to tones 1-6, averaged across root mean square values of all gradiometers and across samples within the a priori mismatch negativity window from 140ms to 160ms; top panel - for baseline controls in blue and patients in orange; bottom panel - for patients at baseline in dark blue and follow-up in light blue. (B) Average mismatch negativity waveform of repetition 1 minus deviant tones; top panel - for baseline controls in blue and patients in orange; bottom panel - for patients at baseline in dark blue and follow-up in light blue. (C) Absolute intraclass correlation for the mismatch negativity calculated using the test-retest data. MCI/AD, mild cognitive impairment or Alzheimer’s disease; MMN, mismatch negativity
The patients’ mismatch response was further attenuated (less negative) at annual follow up compared to patients’ baseline (T = −2.72, p = .005, Figure 1B lower panel). In the longitudinal repeated-measures ANOVA, there was a significant main effect of tone repetition (F(5, 155) = 20.0, p<.001) and progression (baseline versus follow-up, F(1, 31) = 75.2, p < .001), which interacted with each other (F(5, 155)=16.6, p<.001). The mismatch negativity amplitude had high reliability across the baseline and two-week test-retest sessions (ICC = 0.95, p < .001, Figure 1C).
Dynamic causal models
The evoked responses generated by dynamic causal models were accurate, with an average Pearson correlation between generated and observed responses of 0.88 (±1.70) and 0.84 (±0.15) for patients at baseline and follow-up, respectively; and 0.85 (±0.18) for controls at baseline (see the supplementary figures for individual model fits).
The model with the highest model evidence was the same for (1) models across baseline patient and control groups (see Figure 2C) and (2) models across baseline and follow-up scans for patients (see Figure 2D). The winning model identified the inferior frontal cortex as above the parietal cortex in the network hierarchy. The winning model had the combination of exponential and phasic basis functions changes in parameters over successive repetitions, in accordance with (Garrido et al., 2009; Rosch et al., 2019) and in line with a predictive coding framework for the mismatch negativity task rather than stimulus-specific adaptation only (Garrido et al., 2009).

(A) The canonical microcircuit model for evoked responses; (B) Model space for determining the connectivity of the inferior parietal nodes; (C) The free energy (top panel) and posterior probability (bottom panel) of models for the patient and control groups; (D) and for the patient baseline and follow-up groups. Sup, superficial; stell., stellate; inter., interneurons; inhib., inhibitory; F, free energy; Pp, posterior probability; l, left; r, right; A1, primary auditory cortex; STG, superior temporal gyrus; IFG, inferior frontal gyrus; IPC, inferior parietal cortex
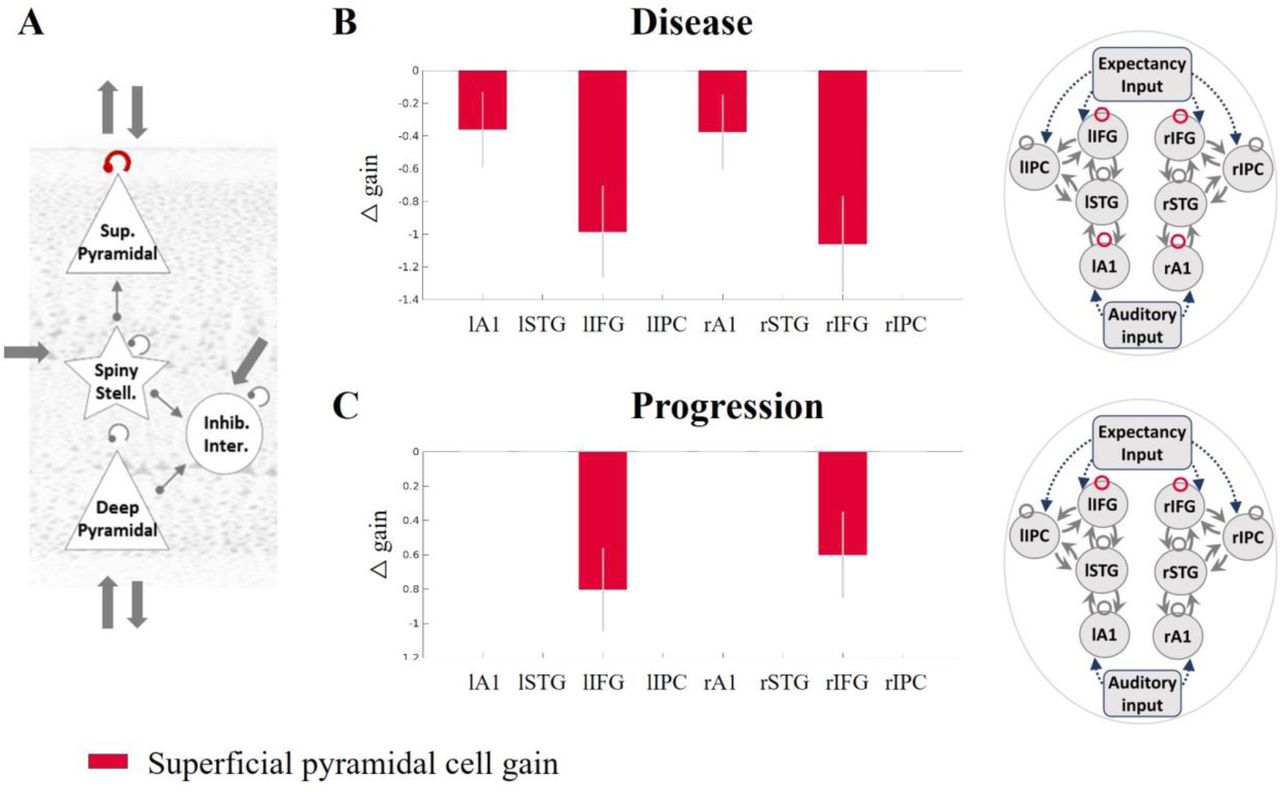
Superficial pyramidal cell gain modulation is reduced by Alzheimer’s disease
The analyses across all regions confirmed that Alzheimer’s disease (Figure 3B: control versus patient PEB model) reduced the gain modulation of superficial pyramidal cells in auditory cortices and inferior frontal gyri. Disease progression also negatively modulated the gain of superficial pyramidal cell self-inhibition in the inferior frontal cortex (Figure 3C: patient baseline versus follow-up PEB model).

(A) The superficial pyramidal cell self-inhibitory connection is shown in red. Regions where this connection was modulated with a posterior probability >0.95 by (B) disease presence and (C) progression are shown in the in red. Sup, superficial; stell., stellate; inter., interneurons; inhib., inhibitory; F, free energy; Pp, posterior probability; l, left; r, right; A1, primary auditory cortex; STG, superior temporal gyrus; IFG, inferior frontal gyrus; IPC, inferior parietal cortex
Extrinsic connectivity: effect of disease and progression
The analyses assessing extrinsic connectivity between superficial and deep pyramidal cells confirmed that the effect of disease was to weaken forward and backward connections. Forward connections were reduced from superior temporal to inferior frontal and inferior parietal cortices and from inferior parietal to inferior frontal cortices; backward connections were reduced from inferior frontal to inferior parietal cortices (Figure 4B). The effect of disease progression was also to reduce the forward connections from superior temporal to inferior frontal cortices and from inferior parietal to inferior frontal cortices and also to reduce backward connections from inferior frontal to inferior parietal cortices (Figure 4C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) The extrinsic connections from superficial pyramidal to deep pyramidal cells (green) and deep to superficial pyramidal cells (blue) are shown in the left hand diagram. Regions between which these connections were modulated with a posterior probability >0.95 are shown in green or blue for (B) disease: patient vs controls and (C) progression: baseline vs follow-up. Sup, superficial; stell., stellate; inter., interneurons; inhib., inhibitory; F, free energy; Pp, posterior probability; l, left; r, right; A1, primary auditory cortex; STG, superior temporal gyrus; IFG, inferior frontal gyrus; IPC, inferior parietal cortex
Discussion
This study confirms that magnetoencephalography of a simple auditory paradigm is sensitive to the presence of Alzheimer’s disease and its progression over 16 months. Furthermore, the mismatch negativity amplitude was highly reliable. Dynamic causal models accurately reproduced scalp responses to the mismatch negativity task. These models revealed the effect of Alzheimer’s disease on laminar and cellular mechanisms. Critically, we confirmed the hypothesis that the presence and progression of Alzheimer’s disease change both the gain modulation of superficial pyramidal cells and extrinsic connectivity between pyramidal cells thereby reducing the mismatch negativity response compared to controls and with disease progression. We also showed reductions in the fast phasic response of intrinsic connections (see supplementary material analysis) to tone repetition, illustrating an effect of Alzheimer’s disease on short-term plasticity.
We propose the mismatch negativity response as an early neurocognitive marker of Alzheimer’s disease. Previous mismatch negativity studies have also shown differences between healthy older adults and people with mild cognitive impairment (Ji et al., 2015; Papadaniil et al., 2016; Gao et al., 2018; Laptinskaya et al., 2018) and Alzheimer’s disease (Savolainen et al., 2001; Pekkonen et al., 2005; Cheng et al., 2012; Hsiao et al., 2014; Papadaniil et al., 2016; Jiang et al., 2017). Although the mismatch paradigm does not directly probe episodic memory, it is correlated with episodic memory in people at risk of Alzheimer’s disease (Gao et al., 2018; Laptinskaya et al., 2018) and the mismatch response is reduced by fibrillary amyloid-beta in preclinical models (Kim et al., 2020). The mismatch paradigm has important advantages as a potential platform in translational studies and for drug development. For example, it requires minimal training, can be undertaken at any stage of disease, has a direct animal task homologue, and it avoids the ambiguity in interpreting tasks on which memory performance differs between groups. The task-induced physiological changes over successive tone repetitions, including the deviant tone, reflect short-term plasticity in cortical microcircuits. Changes to short-term plasticity are an early feature of preclinical models of Alzheimer’s disease (Lee et al., 2012), occurring prior to cell death (Selkoe, 2002) and in proportion to cognitive impairment (Chakroborty et al., 2019). The auditory mismatch is also particularly suitable for MEG, since MEG is more sensitive to superficial cortical regions than deep-brain regions, such as structures in the medial temporal lobes.
In the context of the current task, we consider the short-term plasticity to be the basis of the adaptation of hierarchical neural beliefs about the causes of sensory experience, following a prediction error generated in response to an unexpected tone. Specifically, the magnitude of the mismatch response indexes the precision (certainty) of the prediction error (Garrido et al., 2009). As proposed by Garrido and colleagues, 2009, the high precision ‘surprise’ in response to a deviant tone is followed by an initial reduction of precision which gradually recovers as a new standard tone is learnt. This sequence of events leads to the combined phasic-plus-exponential-decay pattern of the magnitude of responses over successive tones (see Figure 2).
For neurophysiological measures and their biophysical models to be useful in support of experimental medicine studies they must do more than merely differ between people with dementia and healthy adults. They should also be sensitive to disease progression and be interpretable in terms of pathological or pharmacological aspects of disease. In this study we focussed on the hypothesised impact of disease on superficial pyramidal cell dynamics and connectivity.
The cholinergic deficit in Alzheimer’s disease is one mechanism by which the cortical generators of the neurophysiological response may differ. For example, cholinergic agonists increase (Moran et al., 2013; Schöbi et al., 2021) and cholinergic antagonists decrease (Schöbi et al., 2021) parameters for the gain modulation of superficial pyramidal cells in the auditory cortex. Moreover. mismatch negativity responses are altered by cholinergic antagonists (Pekkonen et al., 2005) and deep brain stimulation of the nucleus basalis of Meynert, a major source of cholinergic innervation of the cortex, stimulation of which has been shown to affect repetition suppression (Dürschmid et al., 2020). Based on the contrasting dose-related effects of cholinergic agonists and antagonists, and the pyramidal cell vulnerability to neurodegeneration in Alzheimer’s disease, we predicted an effect of disease on the gain modulation of superficial pyramidal cells underlying the abnormal mismatch negativity response. Indeed, Alzheimer’s disease reduced this gain modulation in primary auditory and inferior frontal cortices (see Figure 3), where there is a high degree of cholinergic fibre loss (Geula and Mesulam, 1996). The inferior frontal cortex was situated at the top of the cortical hierarchy of our model and received expectancy input from external regions, such as the hippocampus, which may contribute to the encoding of the precision of prediction errors with cholinergic regulation of the region at the top of a processing hierarchy (see Figure 3, Barron et al., 2020, and Figure 2, current study).
In addition to the changes of gain modulation within the canonical microcircuit of temporal and frontal regions, we illustrate connectivity changes between regions in our large-scale network of temporal, parietal, and frontal sources (Figure 4). Connectivity changes in large-scale networks related to Alzheimer’s disease have been observed with both fMRI and M/EEG (Yu et al., 2021). For example, Alzheimer’s disease pathology has been associated with changes to functional connectivity of the default-mode network and/or frontoparietal network (Engels et al., 2015; Klaassens et al., 2017; Mandal et al., 2018; Zhao et al., 2019; Wales and Leung, 2021; Yang et al., 2021; Fathian et al., 2022; Moffat et al., 2022; Zhukovsky et al., 2023) and networks responding in mismatch paradigms (Başar et al., 2017; Jovicich et al., 2019; Nguyen et al., 2019; Cope et al., 2022; Gjini et al., 2022).
Pyramidal cells are the principal source of such inter-regional connectivity. Our model-based analysis suggests that pyramidal cell connectivity is impaired by the presence and progression of Alzheimer’s disease (see Figure 3 and Figure 4). Toxic species of aggregated Tau affect pyramidal-cell dendritic complexity (Braak and Braak, 1991; Merino-Serrais et al., 2013; Braak and Tredici, 2018) even before axonal aggregation (Braak and Tredici, 2018). Tau oligomers and beta-amyloid aggregates are also synaptotoxic (Lacor et al., 2007; Mijalkov et al., 2021). This will reduce sensitivity to incoming connections, reducing effective connectivity (Ahnaou et al., 2017).
Another potential contributor to the abnormal mismatch response is synaptic loss and later pyramidal cell death. Transgenic models show disruption of pyramidal cell activity prior to cell death (Lison et al., 2014) with significant degeneration of synapses (Oakley et al., 2006). Human in vivo studies with SV2a-binding PET ligands confirm this loss of synaptic density (Venkataraman et al., 2021). From post-mortem human studies, there is substantial loss of layer 3 (superficial) and layer 5 (deep) pyramidal cells in people with Alzheimer’s disease compared to controls (Hof et al., 1990). This loss is proportional to the burden of tau neurofibrillary tangles (Hof et al., 1990). This may explain the reduced extrinsic connectivity between superficial and deep pyramidal cells we observed (Figure 4).
For extrinsic connections between superficial and deep pyramidal cells, connections to and from the parietal regions were especially affected (Figure 4B and C). The parietal cortex is commonly implicated in functional and structural imaging of Alzheimer’s disease (Wang et al., 2015). In contrast, connections to and from primary auditory cortices were not significantly affected, in accord with the classical progression of pathology over Braak stages (Braak and Tredici, 2018).
Our study has several important limitations. First, our diagnostic groups were defined by clinical diagnoses, albeit supported by amyloid biomarker status determined using PET or cerebrospinal fluid assays. Further work could examine preclinical stages or phenotypic variants. Second, we applied a simplified anatomical model based on earlier studies of the mismatch negativity response augmented by the parietal cortex because of its involvement in Alzheimer’s disease. The model space was defined to suit our hypotheses; however, other models could exist which better explain the data. We did not, for example, include the hippocampus and adjacent medial temporal cortex, which are affected early in the progression of Alzheimer’s disease. However, magnetoencephalographic signals from the hippocampus present methodological and signal-to-nose challenges. Moreover, hippocampal signals may be implicit in the model in the form of the expectancy inputs to the upper layers of the current generative model. A third consideration is the use of concomitant medication in the patient group; however, participants were required to be on a stable dose for at least 30 days prior to participation in the study. The impact of treatments, such as cholinesterase inhibitors, could therefore have moderated the results. However, only 4 patients were on cholinesterase inhibitors (donepezil hydrochloride) in the baseline comparison, 3 of these completed the longitudinal assessment while none in the test-retest group took cholinesterase inhibitors. We would expect such cholinesterase inhibitors to reduce (rather than exacerbate) differences between groups. Fourth, there was a significant difference in age between patient and control groups. However, supplementary PEB analyses which included age as a regressor of non-interest showed similar results (see supplementary analysis). Fifth, attrition at follow-up could affect results. However, the attrition rate of 29% over an average 16-month interval is in line with previous longitudinal studies prior to COVID-19. The original study protocol planned for an interval of 12 months, in accord with many early phase clinical trials of disease-modifying agents. However, the COVID-19 pandemic lockdowns extended the average interval. Future in vivo, human studies could determine whether disease-modifying treatments targeting the cholinergic deficit or tau depositions in pyramidal cells alleviate the reported findings. Future research can use new methods for incorporating information about where in the cortex the synaptic loss is located using dynamic causal modelling (Adams et al., 2023; Jafarian et al., 2023) or post mortem data. The current method would also be applicable to presymptomatic people and further research is needed to determine sensitivity prior to mild cognitive impairment.
In conclusion, we have shown how dynamic causal modelling of human neurophysiological non-invasive recordings is reliable and sensitive to detect the effects of Alzheimer’s disease and progression of Alzheimer’s disease. Specifically, we confirmed changes in the pyramidal cell gain and connectivity as predicted by preclinical and post mortem studies. This methodology helps to bridge the gap between preclinical and clinical studies, identifying mechanistically-informative markers of disease that are sensitive to disease presence and progression.
Data Availability
Imaging data and clinical scores are hosted by Dementias Platform UK Imaging Platform (https://portal.dementiasplatform.uk), using XNAT (https://www.xnat.org). Data will be made available with a managed access process through Dementias Platform UK, subject to requesters agreeing to a Code of Conduct to preserve data security, confidentiality and privacy.
Funding
This work is primarily funded by the Dementias Platform UK which is funded by the Medical Research Council (MC_UU_00030/14 & MR/T033371/1), Janssen, AstraZeneca, Araclon, IXICO, Somalogic, GlaxoSmithKline, Invicro, Cambridge Cognition and Cognetivity. The study has additional support from Alzheimer’s Research UK (ARUK-PG2017B-19), the Wellcome Trust (220258), Medical Research Council (SUAG/092 G116788; SUAG/096 G116788), NIHR Cambridge Biomedical Research Centre (NIHR203312) and NIHR Oxford Health Biomedical Research Centre (NIHR203316). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. For the purpose of open access, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Competing interests
MT is an employee from Janssen Research & Development, a Division of Janssen Pharmaceutica NV., Beerse, Belgium, and owns stock or stock options in the company. MP is employed by AstraZeneca and may currently hold AstraZeneca stocks or stock options. SL is employed by Eli Lilly and may currently hold Eli Lilly stock.
Supplementary material
Please see separate word document for supplementary material
Appendix
In this section, we provide further detail on the biologically-informed generative model of the scalp MEG data used in this paper.
Dynamic casual modelling entails a variational Bayes inversion of biologically-informed models to neuroimaging data under the Laplace assumption (that the priors and posteriors of the unknown parameters have a Gaussian distribution) (Friston et al., 2003, 2007, 2019; Kiebel et al., 2007). We use dynamic causal modelling to infer the posterior densities of model parameters and the model’s free energy score from features of the electrophysiological recordings, in this case, the evoked response. The negative free energy of the model provides a lower bound for the log model evidence and is the difference between the predictions of the model’s accuracy and complexity. The objective of variational Laplace is to maximise the free energy score by iteratively updating and adjusting the model parameters given data features and prior information. As the model’s parameters are updated, the accuracy of the predicted model’s response to the observed response is gradually improved, while the inversion scheme penalises model complexity to reduce the risk of overfitting (Zeidman et al., 2023). The model evidence of different models, with neuronal architectures differing according to the hypothesis being tested, can be compared to elucidate the most likely underlying causes of the observed brain response.
In this study, the neuronal model of each source is the convolution canonical microcircuit model (spm_fx_cmc.m). This model compromises an intrinsic anatomical network in each region, with inhibitory interneurons, superficial and deep pyramidal cells and spiny stellate excitatory populations as shown in Figure 2A. Specification of the parameters and their priors are set at the default values (spm_cmm_nmda_priors.m). The mean, presynaptic firing rate of each population is scaled by extrinsic connectivity parameters and convolved with a synaptic impulse response function (modelled by single time constant, T, with an alpha-shaped kernel,  to produce the average membrane potential. The mean firing rate (average action potentials) is the sigmoid transformation (denote by σ) of the membrane potential. Mathematically, the membrane potential in a population j within region i,
to produce the average membrane potential. The mean firing rate (average action potentials) is the sigmoid transformation (denote by σ) of the membrane potential. Mathematically, the membrane potential in a population j within region i,  , can be calculated by convolving (denoted by ⊗) the presynaptic firing rate (the sum of all intrinsic, extrinsic, and experimental firing inputs) with a synaptic kernel as follows:
, can be calculated by convolving (denoted by ⊗) the presynaptic firing rate (the sum of all intrinsic, extrinsic, and experimental firing inputs) with a synaptic kernel as follows:
 In equation 1, intrinsic connections
In equation 1, intrinsic connections  (within region j, and from population k to i) are specified according to laminar-specific features of cortical columns. Each population is subject to self-inhibition, a proxy of self-regulation, to ensure the stability of the model. Extrinsic connectivity, denoted in equation 1 as
(within region j, and from population k to i) are specified according to laminar-specific features of cortical columns. Each population is subject to self-inhibition, a proxy of self-regulation, to ensure the stability of the model. Extrinsic connectivity, denoted in equation 1 as  (from distal source k to i and from population l to j), can be bottom-up (originating from superficial pyramidal cells in a lower level and targeting spiny stellate cells and deep pyramidal cells in a higher level of a cortical hierarchy) and/or top-down (from deep pyramidal cells at high cortical levels to inhibitory interneurons and superficial pyramidal cells in the lower levels; (elleman and Van Essen, 1991; Hilgetag et al., 2000). A thalamic drive is denoted by um, which excites spiny stellate cells (in some sources) and models the effect of experimental input (scaled by C). The model of an experimental input is a Gaussian function (with latency prior of 70 ± 16 ms).
(from distal source k to i and from population l to j), can be bottom-up (originating from superficial pyramidal cells in a lower level and targeting spiny stellate cells and deep pyramidal cells in a higher level of a cortical hierarchy) and/or top-down (from deep pyramidal cells at high cortical levels to inhibitory interneurons and superficial pyramidal cells in the lower levels; (elleman and Van Essen, 1991; Hilgetag et al., 2000). A thalamic drive is denoted by um, which excites spiny stellate cells (in some sources) and models the effect of experimental input (scaled by C). The model of an experimental input is a Gaussian function (with latency prior of 70 ± 16 ms).
To generate simulated scalp MEG data, we specified a network of interconnected neuronal sources that are active under the experimental paradigm. The activities of the neuronal sources are approximated as equivalent current dipoles, whose orientation is estimated with symmetry constraints as part of the fitting of the whole model. The models were fitted to sensor data which had been reduced using singular value decomposition to eight principal modes for computational expediency. The generative model is thereby able to predict evoked responses to experimental inputs (in this study, auditory inputs). We fitted this generative model to the scalp MEG data using the variational Laplace scheme in the SPM12 software.
References



