
Abstract
Introduction Our knowledge of X-linked Alport Syndrome [AS] comes mostly from selected cohorts with more severe disease.
Methods We examined the phenotypic spectrum of X-linked AS in males and females with a genotype-based approach using data from the Geisinger MyCode DiscovEHR study, an unselected health system-based cohort with exome sequencing and electronic health records. Patients with COL4A5 variants reported as pathogenic (P) or likely pathogenic (LP) in ClinVar, or protein-truncating variants (PTVs), were each matched with up to 5 controls without COL4A3/4/5 variants by sociodemographics, diabetes diagnosis, and year of first outpatient encounter. AS-related phenotypes included dipstick hematuria, bilateral sensorineural hearing loss (BSHL), proteinuria, decreased eGFR, and ESKD.
Results Out of 170,856 patients, there were 29 hemizygous males (mean age 52.0 y [SD 20.0]) and 55 heterozygous females (mean age 59.3 y [SD 18.8]) with a COL4A5 P/LP variant, including 48 with the hypomorphic variant p.Gly624Asp. Overall, penetrance (having any AS phenotypic feature) was highest for non-p.Gly624Asp P/LP variants (males: 94%, females: 85%), intermediate for p.Gly624Asp (males: 77%, females: 69%), compared to controls (males: 32%; females: 50%). The proportion with ESKD was highest for males with P/LP variants (44%), intermediate for males with p.Gly624Asp (15%) and females with P/LP variants (10%), compared to controls (males: 3%, females 2%). Only 47% of individuals with COL4A5 had completed albuminuria screening, and a minority were taking renin-angiotensin aldosterone system (RAAS) inhibitors. Only 38% of males and 16% of females had a known diagnosis of Alport syndrome or thin basement membrane disease.
Conclusion In an unselected cohort, we show increased risks of AS-related phenotypes in men and women compared to matched controls, while showing a wider spectrum of severity than has been described previously and variability by genotype. Future studies are needed to determine whether early genetic diagnosis can improve outcomes in Alport Syndrome.
Introduction
Alport syndrome (AS) is the second most common genetic cause of end stage kidney disease behind polycystic kidney disease, affecting approximately one in 2320 individuals1,2. Nearly 85% of total cases of AS in the past have been due to variants in the COL4A5 gene found on the X chromosome, with the rest of individuals with AS having variants in the COL4A3 and COL4A4 genes responsible for autosomal recessive and dominant inheritance patterns1. Individuals with AS can have phenotypic manifestations related to abnormalities in the basement membranes of the glomerulus, cochlea of the ear, and lens of the eye, resulting in microscopic hematuria, sensorineural hearing loss, and ocular disturbances. Each collagenous IV chain contains a collagenous domain made of Gly Xaa Yaa triplet repeats, resulting in a rigid protein structure3. Thus, glycine missense variants disrupting the intermediate collagenous domains as well as protein-truncating variants (PTVs) are likely to be deleterious. Some but not all studies suggest a worse prognosis for PTVs than missense variants4–8. One glycine missense variant, p.Gly624Asp, is adjacent to a non-collagenous region and known to have a more mild phenotype.9
Although females are more likely to carry COL4A5 variants due to the nature of the X-linked inheritance pattern, males are more severely impacted, with reported risk of progression to ESKD reported at nearly 100% in COL4A5 males compared to 25% in similarly affected females10,11. An important limitation to the existing literature is that participants in these studies have been selected from families known to be affected by AS, likely leading to an overestimation of penetrance and phenotypic severity. As availability of genetic testing increases and costs of testing decrease, there is an urgent need to understand the penetrance and phenotypic spectrum in the general population to inform families about the implications of a genetic diagnosis of AS.
Using a genotype-first approach, we aimed to examine the phenotypic spectrum of X-linked AS due to COL4A5 variants using an unselected health system-based population. We hypothesized that both males and females with COL4A5 variants would have higher risk of kidney phenotypes though the penetrance and phenotypic severity of disease would be lower than previously reported in more selected cohorts.
Methods
Study Population
There were 170,856 research participants in MyCode who had whole exome sequencing data available after quality control. These research participants are patients in the Geisinger Health System at large who consented to participate in the MyCode Community Health Initiative and were sequenced through the Geisinger-Regeneron DiscovEHR collaboration12. This research study was approved by the Geisinger Institutional Review Board. Patients with ClinVar P/LP or PTVs, as defined below, were classified as cases. We queried ClinVar [date accessed 4/24/2023] for COL4A5 (NM_033380.3) variants previously classified as pathogenic (P) or likely pathogenic (LP) by >1 submitter and no conflicts, or conflicting variants with a ratio of ≥6 P/LP to other classifications13. Predicted protein truncating variants (PTVs; frameshift, nonsense, splice) were identified from those called as high impact variants by variant effect predictor v109. All variants underwent manual review to ensure technical confidence and clinical relevance.14 Any variants that did not meet quality thresholds were confirmed in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory using orthogonal technology prior to inclusion.
Phenotypic outcomes
Phenotypic data sources included International Classification of Diseases (ICD) 9 and 10 codes, blood and urine laboratory data, and chart review. ICD codes were used for hypertension, diabetes, hearing loss, bilateral sensorineural hearing loss, AS, focal segmental glomerulosclerosis (FSGS), and ESKD (Supplemental Table 1). We used the 2021 CKD-EPI equation to calculate eGFR, and we imputed eGFR of 5 ml/min/1.73m2 for patients with kidney transplant.15 Hematuria on dipstick was defined as having trace blood or greater on at least half of all urinalyses, excluding urinalyses positive for leukocyte esterase or nitrites.5 Age at time of ESKD was determined using linkage to the United States Renal Data System,16 and if these data were not available, by chart review. Presence of albuminuria was defined as ever having albumin to creatinine ratio (ACR) ≥ 30 mg/g, and presence of proteinuria by dipstick was defined requiring at least 2 dipsticks. Any phenotypic feature of AS was defined as hematuria on urinalysis or ICD code, albuminuria, eGFR <60 ml/min/1.73m2, FSGS ICD code, ESKD ICD code, or bilateral sensorineural hearing loss. Patients with most recent eGFR data at age ≥18 years were categorized into Kidney Disease Improving Global Outcomes (KDIGO) risk categories using estimated glomerular filtration rate (eGFR) and ACR (Supplemental Table 2).5,17 If ACR was not available, urine dipstick protein data was used for KDIGO risk categorization with dipstick protein 1+ twice classified as ACR 30-299 mg/g, and dipstick protein 2+ or greater twice classified as ACR 300+ mg/g. Chart review was performed on all cases to further examine diagnosis of AS or TBMD, kidney biopsies, treatment with angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs), family history, and genetic testing.
Statistical analysis
Patients with COL4A5 P/LP variants were matched to up to 5 controls (patients without COL4A3, COL4A4, and COL4A5 variants at allele frequency<0.01), with exact matching on age (within 1 year) at time of most recent eGFR, sex, race, diabetes diagnosis, and year of first outpatient encounter. We compared characteristics between patients with COL4A5 P/LP variants and matched controls using chi-square tests for categorical outcomes and paired t-tests for continuous outcomes overall, and by sex and variant group. We used Poisson regression models with robust variance as the primary analysis to estimate prevalence ratios to determine AS-related phenotypic outcomes, stratified by sex and genotype (P/LP variant or the known hypomorphic p.Gly624Asp variant), using each subgroups’ matched controls. 18,19 We also conducted time-to-event analyses with ESKD as the outcome using Kaplan-Meier survival curves with age as the time variable. Patients were censored at age of last eGFR value. The proportional hazards assumption was graphically assessed, and Cox proportional hazards models were adjusted for age at time of first eGFR value in sex-stratified analyses. As prior COL4A5 studies have suggested genotype-phenotype correlations with the worst outcomes for nonsense and frameshift variants, intermediate outcomes for splice variants, and milder phenotypes for missense and in-frame deletion variants,6,20,21 we also estimated risk of ESKD using Cox models for nonsense/frameshift variants, splice variants, the hypomorphic p.Gly624Asp variant, and other missense or in-frame deletion variants. We conducted sensitivity analyses, including time-to-event competing-risks regression using the method of Fine and Gray,22 and analyses using Firth logistic regression, including only one individual from each family. We considered p values <0.05 to be significant. Analyses were conducted using STATA/MP 15.1.
Results
Out of the 170,856 patients, there were 29 hemizygous males (mean age 52.0 y [SD 20.0]) and 55 heterozygous females (mean age 59.3 y [SD 18.8]) with a COL4A5 P/LP variant. The most common COL4A5 P/LP variant was p.Gly624Asp (n=48), and there were 17 other unique variants, including 4 other missense or in-frame deletion variants (n=16 individuals), 7 splicing variants (n=13 individuals), 4 frameshift variants (n=4 individuals), and 2 stop-gained variants (n=3 individuals). Of the 84 individuals with COL4A5 P/LP variants, the vast majority were matched to 5 controls (78 individuals matched to 5 controls, 2 to 3 controls, 2 to 2 controls, 2 to 1 control). Compared to matched controls, individuals with COL4A5 P/LP variants were more likely to have urinalysis (96% vs 82%, p=0.001) and ACR (47% vs 26%, p<0.001) results available in addition to having a hypertension diagnosis (68% vs 51%, p=0.004) whereas 100% of patients in both groups had evaluation of eGFR (Table 1). The median (interquartile interval) year of first outpatient visit was 2003.5 (2001-2009.5) for individuals with COL4A5 and 2004 (2001-2009) for matched controls.
Prevalence of AS Phenotypes by Sex
Overall, males with COL4A5 P/LP variants had the highest risk of AS-related phenotypes, followed by males with the hypomorphic p.Gly624Asp variant and females with COL4A5 P/LP variants, with lowest risk in females with p.Gly624Asp (Table 2, Figure 1). The p.Gly624Asp variant was associated with a milder phenotype than other P/LP variants for both males and females. Males with p.Gly624Asp demonstrated lower penetrance (n=13; 0% AS ICD, 15% ESKD ICD, 31% hearing loss ICD, 23% eGFR <30 ml/min/1.73m2) compared to other hemizygous COL4A5 P/LP males (n=16; 31% AS ICD, 44% ESKD ICD, 50% hearing loss ICD, 44% eGFR <30 ml/min/1.73m2). KDIGO risk categorization demonstrated that 33% of p.Gly624Asp males (mean age 55.7 y [SD 17.1] had a high-risk category or greater as compared to 80% of other hemizygous COL4A5 P/LP males (mean age 49.0 y [SD 22.2]) (Table 2, Figure 1). Similarly, the prevalence of high-risk KDIGO category or greater in p.Gly624Asp females (mean age 60.3 y [SD 14.8] was 24% compared to 68% of other P/LP females (mean age 59.9 y [SD 18.1]).

KDIGO risk categories created using eGFR and ACR. If ACR not available, then urine dipstick protein data used with dipstick protein 1+ twice classified as ACR 30-299 mg/g, and dipstick protein 2+ or greater twice classified as ACR 300+ mg/g. KDIGO risk categories defined as: 0 - no CKD or hematuria; 1 - hematuria alone (eGFR ≥60, ACR <30); 2 – Moderately increased risk (eGFR 45-59 and ACR<30 or eGFR ≥60 with ACR 30-299); 3 – High risk (eGFR 30-44 with ACR<30 or eGFR 45-59 with ACR 30-299, or eGFR ≥60 with ACR 300+); 4 – Very high risk (eGFR 15-29 with ACR<300 or eGFR 30-44 with ACR ≥30 or eGFR 45-59 with ACR ≥300); 5 – Extremely high risk (eGFR<15 or eGFR 15-29 with ACR 300+; see Supplemental Table 2). Two cases and 6 controls <18 years of age were excluded from this categorization.
All groups had significantly higher risk of AS phenotypic features compared to matched controls with prevalence ratios (PR) of 3.13 (95% CI: 2.18, 4.48) for males with P/LP variants, 2.20 (95% CI: 1.39, 3.48) for males with p.Gly624Asp, 1.75 (95% CI: 1.34, 2.29) for females with P/LP variants, and 1.35 (95% CI: 1.03, 1.76) for females with p.Gly624Asp (Table 3, Figure 2). Risks of the most common AS manifestation, dipstick hematuria, were similar for males with P/LP variants (PR 2.36, 95% CI: 1.07, 5.23), males with p.Gly624Asp (PR 3.34, 95% CI: 1.80, 6.21), females with P/LP variants (PR 2.59, 95% CI: 1.73, 3.87), and females with p.Gly624Asp (PR 1.86, 95% CI: 1.28, 2.70). Males and females with p.Gly624Asp tended to have milder phenotypic presentation than their counterparts with other P/LP variants. For example, prevalence ratios for ESKD were 35.00 (95% CI: 4.57, 268.10) for males with P/LP variants, 3.08 (95% CI: 0.56, 16.80) for males with p.Gly624Asp, 9.30 (95% CI: 0.88, 98.67) for females with P/LP variants, and 1.61 (95% CI: 0.17, 15.07) for females with p.Gly624Asp. Males with P/LP and p.Gly624Asp variants had significantly increased risk of hearing loss ICD diagnosis whereas females with P/LP variants or p.Gly624Asp did not. Sensitivity analyses restricted to one individual per family showed consistent results (Supplemental Tables 3). Variant-level phenotypic characteristics, stratified by sex, are shown in Supplemental Table 4.

Associations between COL4A5 P/LP variants and renal phenotypes examined using Poisson regression with robust variance in analyses in stratified analyses by genotype and sex.
Time-to-event Analyses for ESKD by Genotype
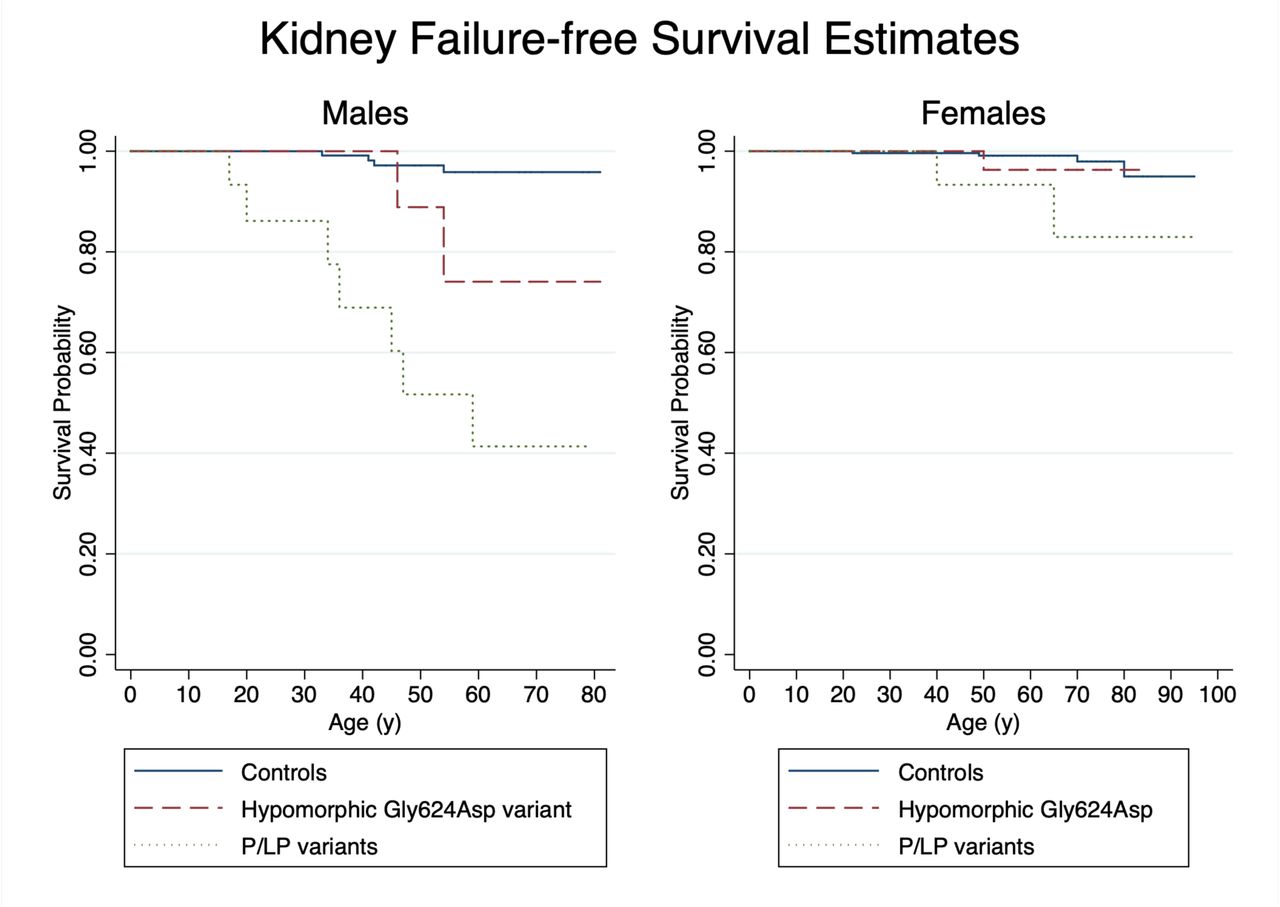
Kaplan-Meier survival curves for ESKD-free survival followed similar patterns (Figure 3). Overall, there was no difference in deaths in patients with COL4A5 variants and controls (7% vs. 7%; p=0.95) with mean age of death of COL4A5 patients 70.8 y (range 54.3-80.7). Table 4 shows hazard ratios (HR) for ESKD were significant for males with P/LP variants (HR 20.14, 95% CI: 5.85, 69.38; p<0.001), females with P/LP variants (HR 5.65, 95% CI: 1.00, 31.75), and of borderline significance for males with the hypomorphic p.Gly624Asp variant (HR 5.38, 95% CI: 0.97, 29.75). Females with the hypomorphic p.Gly624Asp variant were not at significantly increased risk of ESKD (HR 1.95, 95% CI: 0.22, 17.59). When examined by additional variant types, (e.g. nonsense/frameshift variants, splice, hypomorphic p.Gly624Asp, other missense or in-frame), risk was similar between variant groups though 95% intervals were very wide due to small sample sizes (Supplemental Table 5). Results accounting for competing risks of death were consistent (Supplemental Table 6). Mean age of ESKD onset in males was 36.9 y (SD 15.0) males with P/LP variants and 50.0 (SD 5.7) for males with Gly624Asp. Mean age of ESKD onset was 51.7 (SD 12.6) for females with no difference between those with P/LP or Gly624Asp variants.

{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier curves comparing risk of ESKD by genotype for females and males with age as the time variable.
Individuals with a COL4A5 P/LP variant and a second COL4A3/4/5 variant
There were 15 individuals who had an additional rare (AF<0.01) variant in COL4A3/4/5 with 4 (27%) having ESKD (Supplemental Table 7). The individuals with ESKD included a male with COL4A5 splice variant c.438+2T>G and COL4A4 p.Pro352Leu; a male with COL4A5 p.Leu1655Arg and COL4A3 p.Val950Ile variant; a male with COL4A5 p.Leu1655Arg and COL4A3 p.Thr1489Ile, and a female with COL4A5 c.4964T>G and COL4A3 p.Ile1659Val.
Diagnosis of AS
AS or thin basement membrane disease was documented in only 10/16 (63%) of males with P/LP variants, 1/13 (8%) of males with p.Gly624Asp, 4/20 (20%) of females with P/LP variants, and 4/35 (11%) of females with p.Gly624Asp. A total of 17 individuals (20%) had documentation of a native kidney biopsy performed, and none had undergone a skin biopsy to diagnose AS. Of those with native kidney biopsies, 9 (53%) had a biopsy diagnosis of AS, hereditary nephritis or thin basement membrane disease; FSGS was reported in 6 (33%) biopsies (Table 5). One patient had 2 biopsies as a child that were inconclusive (1st biopsy: insufficient sample, thin basement membrane disease; 2nd biopsy: IgM nephropathy, FSGS) and has yet to be diagnosed with AS. Only 2 (12%) had specific mention of staining for the alpha5 chain of type IV collagen. In terms of genetic diagnosis, only 5 (6%) received genetic testing and 2 (2%) additional patients reported having a family member who had genetic testing confirming diagnosis.
Receipt of Recommended Care for AS
Only 40 (47%) of COL4A5 patients had ever had ACR testing completed though 82 (96%) had urinalysis data. Overall, only 27 (32%) of COL4A5 patients were receiving RAAS inhibitors. Among the 37 individuals with ACR ≥30 mg/g or 1+ protein twice on dipstick (and without ESKD), only 18 (49%) were receiving RAAS inhibitors.
Discussion
In this study using an unselected cohort, we provide more representative quantification of risks associated with X-linked AS, by sex and genotype. Risks of ESKD was highest among males with P/LP variants (44%), intermediate for males with p.Gly624Asp (15%) and females with P/LP variants (10%), and not significantly increased for females with p.Gly624Asp (3%). Overall, we show a wider spectrum of disease severity in both men and women with COL4A5 variants than described in earlier studies that focused on families known to be afflicted by the disease. For example, prior studies suggested males with X-linked AS having a 100% chance of progression to ESKD10. In our cohort, 80% of COL4A5 P/LP males and 33% of COL4A5 p.Gly624Asp males were categorized as having KDIGO high-risk category or greater, and there were 2 men above the age of 70 years who had eGFR ≥60 ml/min/1.73m2. This finding of more variable phenotypic severity has important implications in terms of providing more accurate information for families. The broader phenotypic spectrum using a genotype-first approach in an unselected cohort is consistent with other similar genotype-based studies for conditions ranging from autosomal dominant polycystic kidney disease to monogenic diabetes.5,23,24 We also uncovered gaps in care related to AS. Despite a median follow up time of approximately 14 years, allowing ample opportunity for various forms of testing, less than half of individuals had albuminuria screening completed and very few individuals with COL4A5 had a diagnosis of AS.
With our genotype-based approach, we are also able to study genotype-phenotype correlations in a less biased fashion than prior studies. Prior studies have suggested that nonsense/frameshift variants confer the worst prognosis, followed by splice variants, and then the mildest phenotypes for missense and in-frame deletion variants.6,20 In our study, we did not observe this genotype-phenotype correlation with similar observed risk for missense variants and PTVs. However, numbers were limited for these analyses, and it is possible that lumping variants into various groups may not reflect differential severity of individual variants within each group. Beyond p.Gly624Asp, which was associated with better outcomes for both males and females, we were unable to make firm conclusions about individual variants. Verifying genotype-phenotype correlations will require larger, additional cohort studies with unselected populations.
Our study and others have shown that p.Gly624Asp, thought to have originated in Central and Eastern Europe due to a founder effect between the 12th and 13th century, confers a milder form of disease and clinical course which may be explained by the location of this variant within the type IV collagen gene25,26. The p.Gly624Asp variant is found adjacent to a non-collagenous interruption within the collagenous domain. It has been hypothesized that since p.Gly624Asp occurs adjacent to such an interruption, it likely increases the flexibility of an already flexible region, explaining why it results in a milder form of disease, as compared to variants inducing flexibility in the more rigid parts of the collagenous domain26. However, despite being associated with a milder presentation, males with p.Gly624Asp variants still had a 5-fold increased risk of developing eGFR<30 ml/min/1.73m2, and 15% had ESKD.
Females are often considered by clinicians as simply being “carriers” of X-linked AS, and we found documentation of this in our chart review of this cohort. Although the manifestations of disease are on average less severe as compared to males, significant kidney risks are still present. An earlier study of families with X-linked AS by Jais et al. had reported a 30% risk of ESKD in females with COL4A5 by the age of 60, with higher risk in those with concurrent proteinuria and hearing loss11. In our study, females with P/LP variants tended to be at increased risk of ESKD with 10% developing ESKD by the age of 65y, likely due to the unselected nature of our cohort. Regardless, the diagnosis of AS in women has important implications for affected women as well as for reproductive decision making and family planning.
Prior research by Gross et al. has demonstrated a role for RAAS inhibitors in delaying onset of renal failure and improving life expectancy in AS27. Their study largely focused on AS in males with sibling pairs showing a significantly delayed onset of renal failure in the younger sibling thought to be due to earlier diagnosis and initiation of medical therapy. This delayed onset of renal failure with RAAS blockade was similarly seen in a study examining females with AS28. Even in males and females with hypomorphic Gly624Asp variants, RAAS inhibitors have been shown to be associated with reduced risk of end stage renal failure in an observational study by Boeckhaus et al29. Despite these studies, only 49% of patients were on RAAS inhibitors even when albuminuria was detected on laboratory studies, emphasizing the importance of early genetic testing and screening to identify such patients. This finding raises the question of whether or not diagnosing these individuals with AS would result in better risk awareness leading to improved adherence to medical therapy, and potentially better patient outcomes.
The major strength of our study is the utilization of an unselected health population and exome sequencing data to inform our diagnosis of patients with X-linked AS, allowing for an in-depth exploration of the phenotypic spectrum of disease. Additionally, due to the availability of extensive longitudinal EHR data, we were able to examine a wide breadth of different phenotypic outcomes. However, EHR data was limited for traits such as ophthalmologic and audiometry data. Another limitation of our study is that all patients with COL4A5 in our cohort were of European ancestry. In conclusion, we were able to show in a large, unselected cohort, the wide spectrum of phenotypic presentation of males and females with COL4A5 variants. X-linked AS was underdiagnosed in both males and females with opportunities to improve monitoring of disease progression and better use of guideline-recommended RAAS inhibitors. Further studies are needed to determine whether early genetic diagnosis can improve outcomes in AS.
Disclosures
A.C. has served as a consultant for Novartis, Reata, and Amgen, and has received research funding from Novartis, Novo Nordisk, and Bayer.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Data files. Additional information for reproducing the results described in the article is available upon reasonable request and subject to a data use agreement.
Author contributions
Study design (ARC, HLK), data collection (MZ, KS, KR, BM, IDB, TM), analysis (ARC), interpretation of results (MZ, IDB, TM, ARC), drafted manuscript (MZ, ARC), revised critically for important intellectual content (all authors). All authors have approved the version to be published.
Conflicts of interest
No potential conflicts of interest relevant to this article were reported.
Acknowledgements
We are grateful to the many patients who contributed to the MyCode Community Health Initiative by providing genomic and electronic health information. We thank the Regeneron Genetics Center for providing funding for patient enrollment and exome sequencing for the DiscovEHR study, and we would like to acknowledge the Geisinger-Regeneron DiscovEHR Collaboration for the genotypic and phenotypic data. This work was supported by NIGMS grant GM111913 to T.M.
We thank the USRDS for their support. The data reported here have been supplied by the United States Renal Data System (USRDS). The interpretation and reporting of these data are the responsibility of the author(s) and in no way should be seen as an official policy or interpretation of the U.S. government.
Footnotes
Some additional validation of variants was done, and thus, the numbers are updated. HRs by genotype are shown for P/LP variants and Gly624Asp in the main manuscript now with the smaller subgroups moved to the supplement.
References



